Review - DOI:10.33594/000000827
Accepted 23 September, 2025 - Published online : 10 November, 2025
1National Institute for Physiological Sciences, Okazaki, Japan,
2Department of Integrative Physiology, Graduate School of Medicine, Akita University, Akita, Japan,
3Department of Physiology, School of Medicine, Aichi Medical University, Nagakute, Japan,
4Institute of Biophysics and Biochemistry, National University of Uzbekistan, Tashkent, Uzbekistan,
5Laboratory for Membrane Transport and Biopharmaceutics, Faculty of Pharmacy, Takasaki University of Health and Welfare, Takasaki, Japan
Solute carrier organic anion transporter family member 2A1 (SLCO2A1) is known to be a molecule responsible for prostaglandin transporter (PGT). Recently, SLCO2A1 was, in addition, identified as the core (pore-forming) molecule of large-conductance maxi-anion channel (Maxi-Cl) that is activated by cell swelling, is sensitive to Gd3+, and has a large pore size mediating ATP release. Thus, this protein exhibits transporter/channel dual functions. In the same cell types, SLCO2A1 molecules were found to simultaneously exhibit both PGT and Maxi-Cl activities in a manner independent of each other. Thus, it is suggested that there exist two groups of SLCO2A1 molecules, in parallel, the activation mechanisms of which are differently regulated. After making genome-wide survey for the molecules involved in the activation mechanism of Maxi-Cl/SLCO2A1, the ANXA2-S100A10 complex was identified as its regulatory subunit by conferring protein tyrosine dephosphorylation and cytosolic Ca2+ dependence thereon. In contrast, SLCO2A1 was shown to exhibit PGT activity by physically interacting with ANXA2 alone. In addition, decreased expression of S100A10 was found to augment plasmalemmal SLCO2A1 expression, thereby augmenting PGT/SLCO2A1 activity. In conclusion, current evidence indicates that SLCO2A1 can exert dual functions as ANXA2-S100A10-associated Maxi-Cl activity and S100A10-lacking ANXA2-associated PGT activity in a manner dependent on the S100A10 expression level. From this standpoint, future studies are warranted to be performed on the three-dimensional conformation changes between PGT and Maxi-Cl and physiological/pathological relevance of SLCO2A1.
The large-conductance maxi-anion channel (Maxi-Cl) was functionally discovered in 1983 by Blatz & Magleby [1] in rat skeletal muscle cells in primary culture and was thereafter shown to be expressed in a wide variety of cell types [2]. This channel is activated by cell swelling, ischemic/hypoxic stimulation, and membrane excision and exhibits phenotypical properties [3, 4] which are distinct from those of another swelling-activated anion channel VSOR/VRAC [5, 6]. The most prominent property is its largest single-channel conductance (300-500 pS) among known anion channel types in mammalian cells, including Ca2+-activated CaCC (0.45-1.2 pS), voltage-gated ClC-2 (2-3 pS), acid-activated ASOR/PAC (4-7 pS), cAMP/PKA-activated CFTR (6-12 pS), and cell swelling-activated VSOR/VRAC (10-90 pS) [4]. Maxi-Cl exhibits other phenotypical properties such as high anion-to-cation selectivity with low-field anion selectivity (I– > Br– > Cl–), linear and symmetrical (ohmic) current-voltage relationship, voltage-dependent inactivation kinetics at large positive and negative membrane potentials, marked sensitivity to extracellular Gd3+, and intracellular ATP sensitivity [3, 4].
Although the phenotypical properties were well elucidated, the molecular identity of Maxi-Cl had not been uncovered for a long time. In 2017, however, its core component was at last identified as a member of the solute carrier organic anion transporter family member 2A1 (SLCO2A1 or OATP2A1), which has 12 transmembrane domains, by applying unbiased genome-wide proteomics LC-MS/MS studies on the Maxi-Cl activity-rich fraction of membrane blebs isolated from mouse mammary C127 cells and thereafter by making targeted siRNA/miRNA screening among the selected 15 genes of transmembrane proteins [7].
Since SLCO2A1 has been known as a prostaglandin (PG) transporter [8, 9], this protein exhibits transporter/channel dual-use behavior [7, 10]. To exhibit dual functions, some molecular activation mechanism(s) for SLCO2A1 activities may be different between Maxi-Cl channel and PG transporter (PGT). Not only different but also common regulatory mechanisms were clarified by our more recent studies [11, 12]. Here, we summarize the dual functions of SLCO2A1 and their molecular regulation mechanisms.
The conclusion drawn by genome-wide proteomics approaches that SLCO2A1 is the core molecule of Maxi-Cl was verified by observing the eliminating effect of SLCO2A1 gene knockout on Maxi-Cl activity in mouse C127 cells, the eliciting effect of its overexpression on Maxi-Cl activity in endogenously Maxi-Cl-deficient human HEK293 cells, and the reproducing effect of reconstitution of SLCO2A1 protein on Maxi-Cl activity in the lipid bilayer membrane. Furthermore, when a charge-neutralizing K613G or R560N mutant of mouse SLCO2A1 (Fig. 1: blue-circled) was transfected in HEK293 cells, the channel activity became relatively more selective to cations exhibiting a smaller single-channel conductance [7]. In addition, two other mutants, G222R and P219L (Fig. 1: red-circled), were found to be non-functional in the channel activity [7]. Thus, it is evident that SLCO2A1 represents the pore-forming molecule of Maxi-Cl.
The large unitary conductance of Maxi-Cl suggests that it has a wide pore. As expected, the nonelectrolyte partition study showed that the effective pore radius is around 1.3 nm [13], which is much larger than the pore radius of VSOR/VRAC (~0.63 nm) [14]. Since the pore size is large enough to permeate ATP4– and MgATP2–, the effective radii of which are 0.56-0.58 nm [2], Maxi-Cl may serve as a conductive pathway for ATP release. Indeed, this inference was directly evidenced by observing inward ATP4– currents [2, 15-20] and MgATP2– currents [18, 21] mediated by Maxi-Cl channels in a variety of cell types. Maxi-Cl-mediated ATP release was shown to be induced by osmotic cell swelling in C127 cells [12, 15, 16], mouse embryonic fibroblasts (MEFs) [22], mouse fibrosarcoma L929 cells [20], newborn rat ventricular myocytes [18, 23], and mature rat ventricular myocytes [23], and by ischemic stress in rat cardiomyocytes [16] and mouse astrocytes [24] as well as by ischemia-reperfusion in perfused mouse hearts in situ [7, 25]. The ATP release induced by osmotic swelling was shown to be suppressed in vitro not only by Slco2a1 knockdown [7] but also by Slco2a1 knockout (KO) [12] in C127 cells. In addition, the inhibiting effect of Slco2a1 silencing in vivo on the ATP release was demonstrated in Langendorff-perfused mouse hearts subjected to ischemia-reperfusion [7]. Furthermore, swelling-induced ATP release from HEK293 cells was found to be elicited after overexpression with SLCO2A1 [12]. Thus, it is concluded that SLCO2A1 serves as the pore of Maxi-Cl through which cells release ATP in response to cell swelling and ischemic/hypoxic stress.
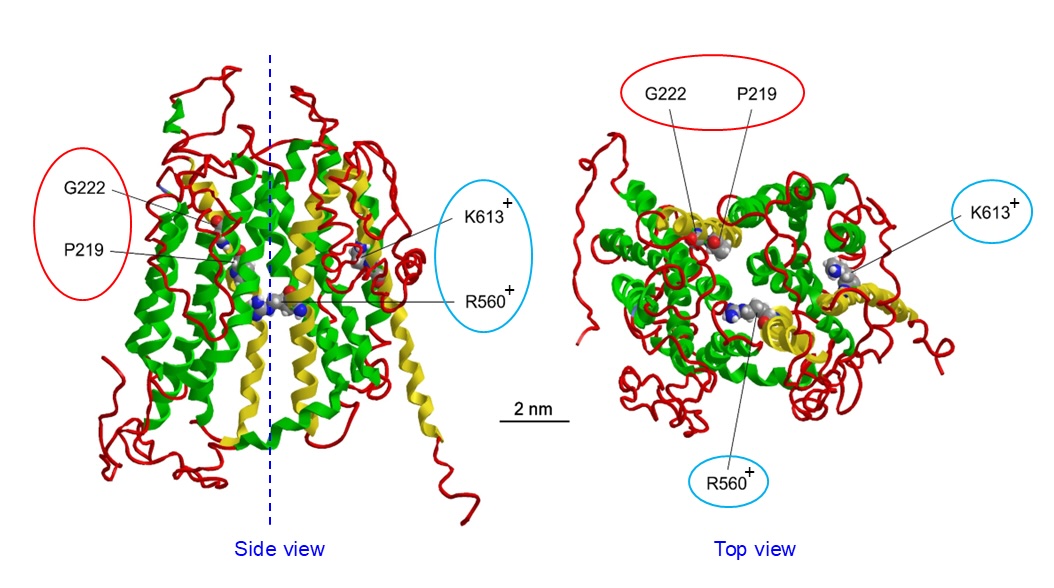
Fig. 1. Contrast Filling. As with the O’Kelly-Marotta scale, the contrast filling of the aneurysm sac is graded: A, complete (>95%); B, incomplete (5-95%); C, neck remnant (<5%); or D, no filling (0%). The left sided drawings show the devices in sidewall aneurysms and the right sided drawings in bifurcation aneurysms.
SLCO2A1 was initially identified as the PG transporter (PGT), which exhibits high affinity particularly for PGE2, PGF2α, and PGD2, by Schuster’s group in rats [8] and humans [26]. In HeLa cells transfected with the SLCO2A1 gene, PGE2 transport was found to be suppressed by depolarization [27] and stimulated by intracellular loading of monovalent anion lactate [28]. PGs are organic anions with pKa values of around 5 and exist in monovalent-charged form at physiological pH [29]. To date, in vitro analyses have demonstrated that PGT/SLCO2A1 mediates cellular PG import as the electrogenic PG/lactate antiporter, presumably exhibiting the 1:2 exchange ratio between PG– and lactate–, as schematically depicted in Fig. 2A. The role of PGT/SLCO2A1 is essential for the termination of extracellular PG signaling by taking up PGs and rapidly inactivating them by 15-hydroxyprostaglandin dehydrogenase in the cytoplasm. Since SLCO2A1 was shown to be expressed on the apical membrane of kidney collecting duct principal cells [30], it is conceivable that SLCO2A1 plays in transepithelial PG reabsorption in the kidney. Supporting this inference, the transepithelial PG transport was experimentally shown to be mediated by SLCO2A1 expressed on the apical membrane, thereby facilitating transepithelial PG transport in monolayer MDCK cells transfected with the rat SLCO2A1 cDNA [31]. More recently, SLCO2A1 was shown to stimulate cellular PG export under inflammatory conditions as well, based on the following observations. First, treatment with an inflammatory factor, lipopolysaccharide (LPS), increased extracellular PGE2 in a manner sensitive to SLCO2A1 gene knockdown in human bronchial epithelial BEAS-2B cells [32]. Second, LPS stimulated PGE2 loading (export) into intracellular acidic compartments (lysosomes) in a manner sensitive to SLCO2A1 gene knockout (KO) in murine macrophage-derived RAW 264 cells and peritoneal macrophages, thereby facilitating exocytotic release of PGE2 from macrophages [33]. Third, H2O2 triggered translocation of SLCO2A1 from the cytosol to the plasma membrane and then induced exocytosis of PGE2 in a manner sensitive to SLCO2A1 gene knockdown in colorectal cancer LoVo cells [34].
In agreement with the fact that SLCO2A1 represents not only Maxi-Cl but also PGT in the same cell types, Slco2a1 KO was shown to simultaneously suppress Gd3+-sensitive ATP release induced by osmotic swelling and PGE2 uptake in C127 cells, whereas SLCO2A1 overexpression in HEK293 cells became induced Gd3+-sensitive ATP release only after osmotic swelling as well as PGE2 uptake under both isotonic and hypotonic conditions [12]. Since PG exhibits a highly hydrophobic nature (with an octanol/water partition coefficient log Po/w of 2.9 [35]), PG transport is likely to be mediated by some transporter (carrier) but not via any channels such as Maxi-Cl, although the size of PGE2 is much smaller than the pore size of Maxi-Cl (Fig. 2B). The ability of PGE2 uptake in C127 cells and HEK293 cells transfected with SLCO2A1 was not affected by hypotonicity-induced cell swelling [12]. Furthermore, in C127 cells under both isotonic and hypotonic conditions, the PGE2 uptake was not affected by the most effective blocker of Maxi-Cl, Gd3+, whereas hypotonicity-induced ATP release was markedly suppressed by Gd3+ [12]. These data indicate that SLCO2A1 serves as the PG transporter in a manner independent of Maxi-Cl activity.
Since SLCO2A1 exhibits dual functions as PGT and Maxi-Cl activities in parallel in a manner independent of each other, it appears that there exists some difference between the regulatory molecular mechanisms for PGT and Maxi-Cl activities.
![Fig. 2. Homology model of three-dimensional mouse SLCO2A1 structure estimated by the l-TASSER algorithm using the crystal structure of glycerol-3-phosphate transporter as a template. Four residues serving as disease-related mutants of mouse SLCO2A1 exhibiting no channel/transporter activities (red-circled) and no transporter but altered channel activities (blue-circled) were included. (modified from [7]).](../images/Fig_68.jpg)
Fig. 2. Homology model of three-dimensional mouse SLCO2A1 structure estimated by the l-TASSER algorithm using the crystal structure of glycerol-3-phosphate transporter as a template. Four residues serving as disease-related mutants of mouse SLCO2A1 exhibiting no channel/transporter activities (red-circled) and no transporter but altered channel activities (blue-circled) were included. (modified from [7]).
Maxi-Cl currents were, for the first time, observed in the excised patches of the plasma membrane from rat skeletal muscle cells in primary culture exposed to ATP-free solution containing free Ca2+ of 0.4 μM [1]. Similar Maxi-Cl currents were then observed to be activated by hypotonic stimulation under the cell-attached conditions in N1E115 neuroblastoma cells [36]. These observations suggest that intracellular high Ca2+ and ATP-deficient conditions are preferable for Maxi-Cl activation.
Cytosolic Ca2+ dependency was indeed qualitatively supported by the observation that application of a Ca2+ ionophore, A23187, activated Maxi-Cl events under the on-cell patch configuration in rabbit cortical collecting duct cells [37] and in Swiss 3T3 fibroblasts [38]. Quantitatively, we demonstrated that unitary Maxi-Cl events evoked by patch membrane excision from C127 cells are enhanced by cytosolic Ca2+ rise in a manner dependent on its free concentration in the range of 0.01 to 10 μM with exhibiting the half activation (Kd) value of 0.515 μM [11].
On the other hand, unitary events of Maxi-Cl were found to become activated by hypoxic or ischemic stress in cell-attached patches on rat ventricular myocytes [18]. This result supports the above inference that a reduction of the intracellular ATP level is favorable for Maxi-Cl activation. The sensitivity of Maxi-Cl activity to cytosolic MgATP was first quantitatively shown by observing the suppressive effect of increased concentrations of intracellular MgATP on Maxi-Cl events with the half inhibitory (IC50) value of 29.2 μM in inside-out patch membranes excised from C127 cells [39]. Then, even in the presence of 0.636 mM MgATP, the Maxi-Cl activity became activated in inside-out patches excised from C127 cells by application of broad-spectrum blockers of tyrosine kinase (genistein and AG18) but not of serine-threonine kinase (H-7, okadaic acid, cyclosporin A, and ascomycin) [39], whereas tyrosine phosphatase blockers (TPhIC, vanadate, and p-bromotetramisole oxalate) inhibited Maxi-Cl events observed in inside-out patch membranes excised from C127 cells and mouse adult fibroblasts (MAFs) [39]. Genistein and AG18 added to bath solution could activate Maxi-Cl currents not only under the cell-attached but also under whole-cell modes in MAF cells even without applying hypotonic and ischemic/hypoxic stimulation [39]. Taken together, it is concluded that Maxi-Cl is activated by tyrosine dephosphorylation in a manner dependent on cytosolic Ca2+ rise.
We started to search for Ca2+-dependent proteins among 686 genes encoding potential membrane-spanning or -associated proteins that are less prominently expressed in Maxi-Cl-deficient C1300 cells compared to those in Maxi-Cl-rich C127 cells by a genome-wide microarray analysis [11]. Four of 12 annexin A (ANXA) subfamilies that are Ca2+-regulated membrane-binding proteins, ANXA1, ANXA2, ANXA3, and ANXA11, were thus selected as first candidates. Among 12 ANXAs, in addition, these four (but not the other eight) proteins were previously shown to be expressed in the bleb membranes of C127 cells by nano-LC-MS/MS proteomics, and, among them, ANXA2 was shown to exhibit the highest value (7.33) of exponentially modified protein abundance index (emPAI) [7]. Then, siRNA-mediated knockdown studies demonstrated that Maxi-Cl activity was markedly suppressed by knockdown of Anxa2, but not Anxa1, Anxa3, and Anxa11 [11]. In addition, hypotonicity-induced ATP release from C127 cells was shown to be markedly suppressed by gene silencing of Anxa2 [11]. These data indicate that ANXA2 regulates Maxi-Cl activity in an augmenting manner.
Plasmalemmal colocalization of ANXA2 and SLCO2A1 was strongly suggested by immunohistochemistry in C127 cells [11]. Moreover, co-immunoprecipitation assay showed that ANXA2 exhibits a physical protein-protein interaction with SLCO2A1 in C127 cells [12] and in HEK293 cells transfected with SLCO2A1 and ANXA2 [11]. Although overexpression of Anxa2 alone in C1300 cells failed to elicit Maxi-Cl activity, this maneuver was found to enhance the Maxi-Cl activity elicited by Slco2a1 overexpression in C1300 cells [11]. These results demonstrate that ANXA2 is involved in the molecular mechanism for activation of SLCO2A1/Maxi-Cl channel.
Since ANXA2 is known to be phosphorylated by protein tyrosine kinases at the residue of Tyr23 [40], there arises a possibility that ANXA2 may confer tyrosine dephosphorylation dependence on Maxi-Cl activity. In accord with this inference, overexpression of a phospho-mimicking ANXA2 mutant, ANXA2-Y23E in which Tyr23 is replaced with glutamic acid, was found to markedly suppress Maxi-Cl activity in C127 cells [11].
ANXA2 is known to form a hetero-tetrameric 2:2 complex with a Ca2+-binding S100 protein family member, S100A10 [41]. Also, Ca2+ affinity of ANXA2 was shown to be enhanced by the complex formation with S100A10 [42]. Therefore, the role of S100A10 (also called p11) in Maxi-Cl activity was next studied, and the following results were obtained [11]. First, microarray analysis showed that S100A10 is more strongly expressed in C127 cells compared to C1300 cells (with a signal ratio of 3.3). Second, gene knockdown of S100a10 suppressed excision-induced Maxi-Cl currents and hypotonicity-induced ATP release in C127 cells. Third, when C127 cells were exposed to the Ac-(1-14) peptide, which has a high affinity for ANXA2 and thereby interferes with the ANXA2-S100A10 complex formation [43], excision-induced activation of Maxi-Cl was prominently inhibited or even sometimes abolished in C127 cells. Fourth, Ca2+-dependent activation of Maxi-Cl was mostly eliminated by S100a10 knockdown in inside-out patch membranes excised from C127 cells. Thus, it appears that the ANXA2-S100A10 complex is responsible for the intracellular Ca2+ dependence of S100A10/Maxi-Cl channel.
Taken together, the ANXA2-S100A10 complex represents the regulatory subunit for Maxi-Cl activity of SLCO2A1 by conferring protein tyrosine dephosphorylation dependence and intracellular Ca2+ dependence on this channel, as schematically depicted in Fig. 3.
![Fig. 3. SLCO2A1 as PG transporter. Four residues serving as disease-related mutants of SLCO2A1 are included. A) Rat SLCO2A1, which consists of 12 transmembrane domains, mediates 1:2 exchange transport (antiport) of one monovalent anion, PGE2, with two monovalent counter anions, mainly lactate. (modified from [62]). B) Function of human/mouse SLCO2A1 as PGT through the physical interaction with ANXA2 that confers protein tyrosine dephosphorylation dependence on this transporter. (modified from [3]).](../images/Fig_67.jpg)
Fig. 3. SLCO2A1 as PG transporter. Four residues serving as disease-related mutants of SLCO2A1 are included. A) Rat SLCO2A1, which consists of 12 transmembrane domains, mediates 1:2 exchange transport (antiport) of one monovalent anion, PGE2, with two monovalent counter anions, mainly lactate. (modified from [62]). B) Function of human/mouse SLCO2A1 as PGT through the physical interaction with ANXA2 that confers protein tyrosine dephosphorylation dependence on this transporter. (modified from [3]).
To search for any differences between the regulatory molecular mechanisms in PGT and Maxi-Cl activities, we then studied the roles of ANXA2 and an obligatory ANXA2 partner protein, S100A10 (p11), in PGT activity by using Anxa2- and S100a10-KO C127 cells and obtained the following results [12]. First, Western blotting study showed that Anxa2 KO induced abolition of S100A10 expression, but S100a10 KO did not alter ANXA2 expression, though the expression of SLCO2A1 was not affected by deletion of Anxa2 or S100a10 in the whole-cell lysates prepared from C127 cells. These observations indicate that S100A10 expression requires ANXA2 expression, but that ANXA2 expression can be maintained even without S100A10 expression, and therefore suggest that the SLCO2A1-ANXA2 and SLCO2A1-ANXA2-S100A10 complexes are separately expressed. Second, in ANXA2-deficient C127 cells, although robust expression of SLCO2A1 was maintained in their plasma membrane fraction, not only swelling-induced ATP release but also PGE2 uptake were prominently reduced. The kinetic analysis showed that PGE2 uptake was uncompetitively inhibited with exhibiting significant decreases in both Km and Vmax values. In contrast, enforced expression of wild-type ANXA2 rescued PGT activity in Anxa2-KO cells by largely restoring both Km and Vmax values. On the other hand, in ANXA2-deficient cells, overexpression of phospho-mimicking ANXA2-Y23E failed to rescue PGT activity, as was the case for Maxi-Cl activity [11], without recovering Km and Vmax values for PGE2 uptake. Thus, it is evident that tyrosine-dephosphorylated ANXA2 is required not only for ATP-releasing Maxi-Cl activity but also for PGT activity. Third, the role of S100A10 in PGT activity was examined. In agreement with suppression of Maxi-Cl currents by S100a10 knockdown in C127 cells [11], hypotonicity-induced ATP release was found to be markedly suppressed in S100a10-KO cells. In contrast, PGE2 uptake was not suppressed but rather enhanced up to around twice by S100a10 KO by increasing the Vmax value alone. In agreement with the known fact that the Vmax value is proportional to the expression of the responsible molecule, plasmalemmal SLCO2A1 expression was found to be increased in S100A10-deficient cells up to more than twice as much. These data show that S100A10 is necessary for Maxi-Cl activity but not for PGT activity of SLCO2A1, and that downregulation of S100A10 upregulates PGT activity of SLCO2A1 by augmenting its plasmalemmal expression, thereby relatively increasing S100A10-lacking ANXA2-associated SLCO2A1/PGT activity compared to ANXA2-S100A10-associated SLCO2A1/Maxi-Cl activity (Fig. 4).
Taken together, it is concluded that ANXA2 alone, but not the ANXA2-S100A10 complex, serves as the regulatory subunit for PGT activity of SLCO2A1 by conferring protein tyrosine dephosphorylation dependence on this transporter, and that diminished S100A10 expression upregulates plasmalemmal SLCO2A1 expression, thereby enhancing PGT activity of SLCO2A1 functioning without S100A10 (Fig. 4).
![Fig. 4. SLCO2A1 as ATP-releasing Maxi-Cl channel. The ANXA2-p11 (S100A10) complex physically interacts with SLCO2A1, thereby forming a hetero-tetramer. Both Ca2+ binding to this hetero-tetramer (either at p11 or ANXA2), triggered by cytosolic Ca2+ rise, and dephosphorylation (at Tyr23 residue) of ANXA2 facilitated by cytosolic ATP fall are prerequisite events for channel activation in response to membrane excision, cell swelling, or ischemia/hypoxia. The channel pore has wide vestibules (the radii of the outer one of 1.42 nm and the inner one of 1.16 nm) and a narrow selectivity filter (the radius of 0.55~0.75 nm), both of which are larger than the radius of ATP. (modified from [3]).](../images/Fig_71.jpg)
Fig. 4. SLCO2A1 as ATP-releasing Maxi-Cl channel. The ANXA2-p11 (S100A10) complex physically interacts with SLCO2A1, thereby forming a hetero-tetramer. Both Ca2+ binding to this hetero-tetramer (either at p11 or ANXA2), triggered by cytosolic Ca2+ rise, and dephosphorylation (at Tyr23 residue) of ANXA2 facilitated by cytosolic ATP fall are prerequisite events for channel activation in response to membrane excision, cell swelling, or ischemia/hypoxia. The channel pore has wide vestibules (the radii of the outer one of 1.42 nm and the inner one of 1.16 nm) and a narrow selectivity filter (the radius of 0.55~0.75 nm), both of which are larger than the radius of ATP. (modified from [3]).
How the SLCO2A1-ANXA2 complex exhibits conformational changes with and without additional interaction with S100A10 remains elusive. Before solving this issue, the three-dimensional structure of SLCO2A1 itself must be elucidated. The spatial structure of mouse SLCO2A1 was first analyzed, as shown in Fig. 1, based on homology modeling using the crystal structure of the glycerol-3-phosphate transporter [7]. In Fig. 5A, the entire mouse SLCO2A1 molecule is divided into the N-terminal (NTD, blue) and C-terminal (CTD, red) domains. This structure looks relatively like an inward-facing conformation of SLCO2A1/PGT. The pore radius calculated along the central axis (purple line in Inset for Fig. 5A) displays two minima, which presumably correspond to two gates located around T346 and F556.
Recently, the cryo-EM structure of the human SLCO2A1/PGT was elucidated [44, 45]. The structures presented in Fig. 5B and Fig. 5C are drawn based on the PDB 8KGW [44] and PDB 9JUQ [45], respectively. In both structures, the human SLCO2A1 adopts an outward-facing state. The pore radius calculated along the central axis of the human SLCO2A1 (green lines in Inset for Figures 5B and 5C) displays a constriction as narrow as that of the mouse model at the position close to the mouse F556 (corresponding amino acid F557 in the human protein). However, this human SLCO2A1/PGT pore is much wider at the second gate near the mouse T346 (T347 in the human protein). Thus, the modeled mouse SLCO2A1 protein is in the state with both intra- and extracellular gates closed, whereas in the outward-facing human cryo-EM structure, the extracellular gate is open but the intracellular gate is closed. Molecular dynamics simulation made for human SLCO2A1 has demonstrated that Cl– but not K+ ions readily diffused into the central cavity around the regions close to positively charged R561 (corresponding to mouse R560) and K614 (corresponding to mouse K613) [44], suggesting that the structure can selectively accumulate anions along the central path. However, on the molecular dynamics movie (Supplementary Movie S1 in [44]), the chloride ions did not pass through the protein, a result consistent with the closed intracellular gate conformation (Fig. 5B). This would suggest that the SLCO2A1 protein in the outward-facing state captured by cryo-EM is not conductive for anions.
Xia et al [45]. showed that PGE2-bound human SLCO2A1 also exhibits an outward-open conformation. However, SLCO2A1-PGE2was found to tend to close the opened extracellular gate (see Supplementary Fig. 9e, f in [45]). They then generated an inward-open model of human SLCO2A1 on the basis of the inward-open structure of OATP1B1 and of the prediction by AlphaFold2, which is an AI system predicting a three-dimensional (3D) protein structure from its amino acid sequence, and found that the salt-bridge interactions contributing to closing the intracellular gate are disrupted (see Fig. 4d in [45]). Since they did not present PDB-coordinates for the whole 3D-structure of human SLCO2A1 thus predicted, we here present such an inward-open model for the 3D-structure of human SLCO2A1 generated by AlphaFold (Q92959 in https://alphafold.ebi.ac.uk/) in Fig. 5D. In the case of AlphaFold SLCO2A1, the extracellular gate is closed, whereas the intracellular one is open, as evidenced by our present calculations of the pore radius along the central axis for Q92959 (red line in Inset for Fig. 5D). We hypothesize that the protein turns to the Maxi-Cl channel state when both gates adopt an open conformation through the interaction with the ANXA2-S100A10 complex. Future studies on the conformational changes of SLCO2A1 between PGT and Maxi-Cl are awaited, hopefully during interaction with ANXA2 and ANXA2 plus S100A10, respectively.
Physiological and pathological roles of SLCO2A1 have so far been investigated through examination of the effects of changes in expression levels of SLCO2A1 gene and protein. First, an essential role of SLCO2A1 in fever generation was shown by the following two studies. In 2003, LPS-induced fever generation in rats was found to be associated with the reduction in Slco2a1 mRNA in the liver and lung [46]. More recently, LPS-induced fever was shown to be associated with increased PGE2 concentration in the hypothalamus interstitial fluid through a micro-dialysis study in global Slco2a1-KO or monocyte/macrophage-specific Slco2a1-KO mice [47]. Second, the role of renal SLCO2A1 in body fluid regulation was proposed in 2008 based on the observation that a high dietary salt intake stimulates transcription of Slco2a1 in the mouse collecting duct principal cells [30]. Third, an involvement of SLCO2A1 in the parturition process was suggested by the observation that its protein expression in the human fetal (amnion, chorion, and decidua) membranes sharply decreases during term labor [48]. In Slco2a1 KO mice, an increase in the extracellular PGE2 concentration in the placenta was found to cause delayed parturition [49]. Fourth, SLCO2A1 upregulation was shown to play an important role in wound healing deficiency associated with diabetes by the following studies. Syeda et al. [50] revealed that hyperglycemia exposure upregulates expression of SLCO2A1 mRNA and protein in human dermal microvascular endothelial cells in vitro and that in vivo SLCO2A1 expression is profoundly upregulated in diabetic patients suffering from defective cutaneous wound healing. Also, Theocharidis et al. [51] demonstrated upregulation of the human SLCO2A1 gene in vascular endothelial cells at the forearm from diabetic foot ulcers. Fifth, enhanced expression of SLCO2A1 mRNA and protein was shown to be associated with ovulation stimulated by chorionic gonadotropin [52]. Sixth, SLCO2A1 downregulation was shown to be causative of the induction of inflammatory processes. Slco2a1-KO mice intratracheally injected with bleomycin exhibited more severe pulmonary fibrosis [53] with increasing mRNA expression of proinflammatory cytokines (TNF-α and IL-1β) and chemokine (CCL5) in bronchoalveolar lavage cells [54]. By applying proteomics approaches, cigarette smoking was found to reduce the expression of SLCO2A1 protein in human lung membranes [55]. Consistently, exposure to cigarette smoke extracts resulted in decreased expression of Slco2a1 mRNA in rat type 1 alveolar epithelial cell-like cells [54] as well as in human colonic LoVo, colorectal Caco-2, and lung mucoepidermoid NCI-H292 cells (56). Furthermore, acute colitis induced by dextran sodium sulfate was found to be exaggerated by global Slco2a1 KO or its macrophage-specific conditional KO in mice with increasing PGE2 in the intestinal lumen and causing NLRP3 inflammasome activation [57]. In contrast, upregulation of SLCO2A1 mRNA was observed in the proximal esophageal mucosa in patients subjected to nonerosive gastroesophageal reflux disease (GERD) [58]. Seventh, possible involvements of SLCO2A1 in gastrointestinal cancer were also reported. In the adenomatous polyposis (APC) mutant model mice, Slco2a1 KO was shown to ameliorate colonic cancer with extending the life span [59]. In contrast, a heterozygous nonsense mutation, G104X, which has a null SLCO2A1 allele in the SLCO2A1 gene, exhibits early-onset colon neoplasia with or without exhibiting digital clubbing [60]. Also, downregulation of the SLCO2A1 gene was detected in the gastric cancer mucosa of Caucasian patients [61]. So far, these physiological and pathological roles of SLCO2A1 have been taken into consideration in relation solely to PGT activity. However, some of these phenomena are indeed difficult to explain solely by the action of PG. Thus, hereafter, studies must be made also from a viewpoint of ATP-releasing Maxi-Cl activity.
A number of genetic human diseases have been demonstrated to be associated with a large variety of SLCO2A1 mutants. Among them, the most well-known genetic disorders are chronic enteropathy associated with SLCO2A1 (CEAS) and primary hypertrophic osteoarthropathy (PHO), also called pachydermoperiostosis (PDP). CEAS is an enteropathy presenting multiple ulcers in the small intestine, chronic bleeding, and protein loss (see Reviews [62, 63]). PHO is a rare genetic disease mainly characterized by digital clubbing, pachyderma (thickening of the facial skin and/or sculp), and periostosis (periosteal new bone formation) (see Reviews [62, 64]) and sometimes endocrine alterations with low IGF-1 and elevated estradiol levels [65]. CEAS and PHO have so far been reported to be associated with 97 different mutations of the SLCO2A1 gene (see Review [62]). Also, some SNPs of the SLCO2A1 gene were reported to determine the intraocular pressure response to eye drops, latanoprost, in glaucoma patients [66] and to be associated with reduced prostate cancer aggressiveness [67]. Patients suffering from such mutations of the SLCO2A1 gene display a variety of symptoms. Therefore, these symptoms caused by SLCO2A1 mutants must hereafter be examined with relevance not only to PGT activity but also to Maxi-Cl activity of SLCO2A1. For example, loss-of-function G219L and G222R mutants associated with PHO and/or CEAS [68, 69] were shown to lack Maxi-Cl activity as well [7]. Thus, altered functions of these disease-causing SLCO2A1 mutants must be evaluated in future studies in relation not only to PG uptake but also to ATP release, especially in gastrointestinal epithelial cells that express massive Maxi-Cl channel activity [70].
![Fig. 5. Dual functions of human/mouse SLCO2A1 controlled by interaction with ANXA2 and S100A10 (p11). Both functions are independently operated because of their distinct difference in sensitivity to hypotonic stimulation and Gd3+ administration. The expression level of S100A10 (p11) may represent the determinant for the transition of SLCO2A1-ANXA2 complex between S100A10-associated Maxi-Cl and S100A10-lacking PGT activities because downregulation of S100A10 (p11) enhanced PGT activity but simultaneously suppressed Maxi-Cl activity. Blue and brown arrows represent activating and inhibiting effects, respectively. (modified from [3]).](../images/Fig_70.jpg)
Fig. 5. Dual functions of human/mouse SLCO2A1 controlled by interaction with ANXA2 and S100A10 (p11). Both functions are independently operated because of their distinct difference in sensitivity to hypotonic stimulation and Gd3+ administration. The expression level of S100A10 (p11) may represent the determinant for the transition of SLCO2A1-ANXA2 complex between S100A10-associated Maxi-Cl and S100A10-lacking PGT activities because downregulation of S100A10 (p11) enhanced PGT activity but simultaneously suppressed Maxi-Cl activity. Blue and brown arrows represent activating and inhibiting effects, respectively. (modified from [3]).
SLCO2A1 is a unique molecule dually responsible for Maxi-Cl channel and PG transporter activities. ANXA2 and S100A10 are critically involved in the regulation of both activities of SLCO2A1. ANXA2 upregulates both activities by exhibiting a physical protein-protein interaction with SLCO2A1. In contrast, S100A10 plays reciprocal roles in these activities: it upregulates Maxi-Cl activity through physical interaction with ANXA2, whereas its expression downregulates PGT activity presumably by shifting the function of the SLCO2A1-ANXA2 complex from PGT to Maxi-Cl. Thus, it is concluded that SLCO2A1 exerts Maxi-Cl activity by interacting with both ANXA2 and S100A10, but PGT activity by interacting with ANXA2 alone, as schematically depicted in Fig. 4.
As summarized in the preceding section, 3D-structures of human SLCO2A1 elucidated by cryo-EM analyses showed that SLCO2A1 exhibits the outward-facing configuration [44, 45], whereas PGE2-bound SLCO2A1 tends to exhibit the inward-facing configuration [45]. In fact, the present pore radius analysis of mouse/human SLCO2A1 visually showed that either the outward gate at around T346/T347 or the inward gate at around F556/F557 opens, as seen in Fig. 5 (Insets). Since SLCO2A1 functions as PGT and Maxi-Cl by interacting with ANXA2 and ANXA2-S100A10, respectively, in the cells, future cryo-EM studies are awaited to be performed during interaction with ANXA2 and ANXA2 plus S100A10 and to thereby clarify how SLCO2A1 exhibits conformational transformation between one-gate-open transporter PGT and two-gate-open channel Maxi-Cl.
Physiological and pathological significance of SLCO2A1 has been assessed by examining the effects of changes in the expression level and those of mutations of SLCO2A1 only with respect to PGT. However, pathological mechanisms of genetic diseases associated with SLCO2A1 mutation may not be explained solely by altered local distribution and action of PGE2 [71]. Since both PG and ATP are important extracellular signals in the living system, future studies are required to examine the effects of altered expression levels and of mutations of dual-functional SLCO2A1 from the viewpoint not only of the changes in PG uptake but also of those in ATP release. Charge-neutralizing R560N and K613G mutants of mouse SLCO2A1 (Fig. 1: blue-circled) are known to impair the PGT function [72] and were shown to exhibit nonselective channel activity distinct from Maxi-Cl [7]. On the other hand, PGT-disrupting P219L and G222R mutants of SLCO2A1 gene (see Fig. 1: red-circled), which are known to be associated with primary hypertrophic osteoarthropathy (PHO) and/or pachydermoperiostosis (PDP) [68], were shown to produce no evident channel activity [7]. Thus, in future studies, it would be worthwhile to examine whether the R560N/R561N and K613G/K614G mutants of mouse/human (or rat) SLCO2A1 can form the complex with ANXA2-S100A10 and whether the P219L and G222R mutants are unable to form the complex with ANXA2. In addition to such studies focused on SLCO2A1 per se, pathological roles of altered expression levels and of mutations in ANXA2 and S100A10 are also expected to be studied in relation to altered PGT and Maxi-Cl activities of SLCO2A1 in the future.
YO wrote the draft, RZS and TN revised it, and YO finalized the manuscript. YO, PZM, RZS, and TN prepared figures. All authors contributed to the article and approved the submitted version.
The authors have no ethical conflicts to disclose.
The authors have no conflicts of interest to declare.No competing interests related to the methods or materials used in this study.