Review - DOI:10.33594/000000631
Accepted 28 April, 2023 - Published online : 26 May, 2023
1Laboratory for Molecular Mechanics of Cell Adhesion, Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica De Chile, Santiago, Chile;
2Laboratory for Mechanobiology of Transforming Systems, Institute for Biological and Medical Engineering, Schools of Engineering, Medicine and Biological Sciences. Pontificia Universidad Católica de Chile, Santiago, Chile
With 270 million infections annually and nearly half a million death a year, shigellosis is a severe intestinal infection caused by bacteria of the Shigella family. Appearance and spread of drug-resistant strains renewed global concerns for public health and finding novel targets for treatment is fast becoming a priority. To this end, invasins are a potentially good candidate. Also called Ipa(s), which is the short for Invasion Plasmid Antigen, invasins play a key role in mediating bacterial invasion and infection of the host cell. Importantly, they have been reported to hijack inbuilt mechanical capability of the host cells such as cell adhesion and active processes mediated by the actin cytoskeleton to enable bacterial ingress into the host cells. IpaA is an invasin of particular interest as it presents three motifs that mimic vinculin binding sites and thus it allows IpaA to interact with vinculin, which is one of the most critical regulators of cellular and tissue mechanics. Using a mechanobiology point-of-view, we aim to provide an overview of Shigella´s infection mechanism, to highlight recently discovered molecular mechanisms of IpaA/vinculin interaction and to finally discuss their consequences for epithelial cell and tissue mechanical homeostasis that may result in the symptomatic outcomes seen in severe shigellosis.
Bacteria of the Shigella family (S. flexneri, S. sonnei, S. dysenteriae, and S. boydii) are responsible for an infective bacterial disease termed shigellosis. As reported by the World Health Organization, shigellosis is estimated to cause 270 million infections annually, mostly concentrated in sub-Saharan Africa and South Asia. It is the major cause of bacillary dysentery with over 446 000 deaths among all ages and it has a great prevalence (between 13 and 30%) in children younger than 5 years [1, 2]. Besides these staggering numbers, shigellosis has been at the center of renewed epidemiological concerns due to the emergence and spread of extensively drug-resistant (XDR) and multidrug-resistant (MDR) bacterial strains, including recent outbreaks in several European countries as reported by the WHO [3]. Bacterial transmission commonly occurs via contaminated food and water or, more rarely, via air-borne or person-to-person contact [4]. Bacteria of this family thrive in the gut microenvironment and use colonic epithelia as point of entry where, by disrupting epithelial integrity and barrier function, they cause the typical symptoms associated with dysentery (e.g., dehydration, fever, abdominal cramps)[5]. In vulnerable subjects such as malnourished individuals, children, and cancer patients, this may evolve into severe pathological scenarios including bloody diarrhoea, bloodstream infection, haemolysis, reactive arthritis, kidney failure and even death [6]. Thus, due to shigellosis´ incidence, severity, and the appearance of XDR and MDR, renewed importance should be given to understand the molecular mechanisms of bacterial invasion machinery and the cellular/tissue consequences of infection in order to develop novel therapeutic interventions. Interestingly, new evidence demonstrated that Shigella´s infection machinery uses ¨molecular mimicry¨ to specifically targets important components responsible to regulate mechanical functions of epithelial cells, such as cell migration and cell-cell adhesion, needed for e.g., wound healing and tissue regeneration [7]. While such strategy may have evolved to facilitate bacterial invasion, it is important to understand the molecular mechanisms underlying host-pathogen interactions, its consequences in terms of disruption of epithelial tissues’ functions and the emerging pathophysiological scenarios. This short review aims to explore these aspects from a molecular and cellular mechanobiology point-of-view.
Epithelial cells form functional tissues and maintain their homeostasis by establishing robust and dynamic adhesions between cells and with the extracellular matrix through specialized protein complexes called adherens junction (AJs) and focal adhesion (FAs), respectively [8–13]. These complexes are responsible to transmit cellular forces generated by the associated actin cytoskeleton and regulate fundamental biological processes (e.g. cell migration, differentiation, and proliferation [14]) and mechanical functions of the tissue (e.g. collective cell migration, wound healing and tissue regenerations) [15–17]. Force transmission between the cytoskeleton of neighbouring cells is mediated at AJs by cadherins whereas at FAs integrins mediate heterophilic adhesion of cells with the extracellular matrix (ECM) via the interaction of the extracellular portion of integrins with ECM proteins. In both cases, a complex network of proteins mechanically connects actin cytoskeleton with the membrane receptor (cadherins or integrins). In both cases, a mechanotransduction module can be found at the core of these protein complexes. This is composed of a protein trimer formed by vinculin, α- and β-catenin at AJs [11, 13, 18] and vinculin, paxillin and talin in FAs [19–21]. Both mechanotransduction modules share remarkable similarities in terms of layered structural organization and mechanisms of force transduction that involve protein conformational changes determined by either biochemical signaling and/or forces to modulate engagement of the structure with the actin cytoskeleton. Such mechanism is commonly referred to as molecular clutch in analogy with the mechanical shaft found in car that allows to differential engage the engine with the tires [11, 20, 22].
Mechanosensitive protein vinculin is a common and crucial node of both AJ and FA complexes where it provides a structural and functional link for force transmission in response to internal and external forces [11, 13, 20, 22, 23]. The crystal structure of vinculin shows (Figure 1A) that the head domain (D1 domain) physically interacts with the tail domain in an autoinhibited conformation [24]. When in this conformation, the binding sites for other components of the force transduction module molecules (namely α-catenin and talin in AJ and FA, respectively) are buried within the D1 and not accessible for binding. Mechanical cues (i.e., tension) or biochemical signaling (i.e., phosphorylation) can force vinculin to open, thus exposing the four α-helices bundle that consequently becomes available for interactions (Figure 1B) [25, 26]. Also, vinculin´s binding partners possessing vinculin binding sites (VBSs) motifs (i.e., α-catenin and talin, in AJ and FA respectively) can directly activate vinculin. The physical interaction in the binding pocket within the vinculin´s four α-helices domain induces an overall distortion of vinculin, that in turn reduces head-to-tail affinity [27]. Furthermore, both α-catenin and talin in their close conformation have their VBSs buried within their structure and, similarly to vinculin, through mechanosensitive activation they can switch to an open conformation, to expose the VBSs, providing a second layer for regulating mechanotransduction [23].
Importantly, the same molecular complexes and mechanisms are used by the cells for mechanosensitive endocytosis and phagocytosis [22]. Thus, it is not surprising to find that various pathogenic microorganisms (i.e., Shigella, Salmonella, Yersinia, E. coli) have evolved mechanisms to invade non-phagocytic cells by targeting the host adhesive mechanisms [30]. At the molecular level this is done by using a set of specialized molecules collectively called invasins (Ipa, Invasion Plasmid Antigen) [31, 32]. This remarkable adaptation facilitates bacterial uptake through mechanisms of ligand mimicry. To mediate invasion, the bacterium uses bacterial ligand recognition motif to interacts with the host receptors and create new adhesive structures with the host. In turn, this may induce the reorganization of the host´s cell adhesion machineries and the associated actin cytoskeleton and ultimately it interferes with tissue homeostasis [33, 34]. Thus, understanding the molecular mechanisms that promote host-pathogen interaction associates with mechanical defect of cell adhesion and, consequently, tissue homeostasis, could provide innovative therapeutic strategies by, e.g., specifically hindering association of invasins with their host target.
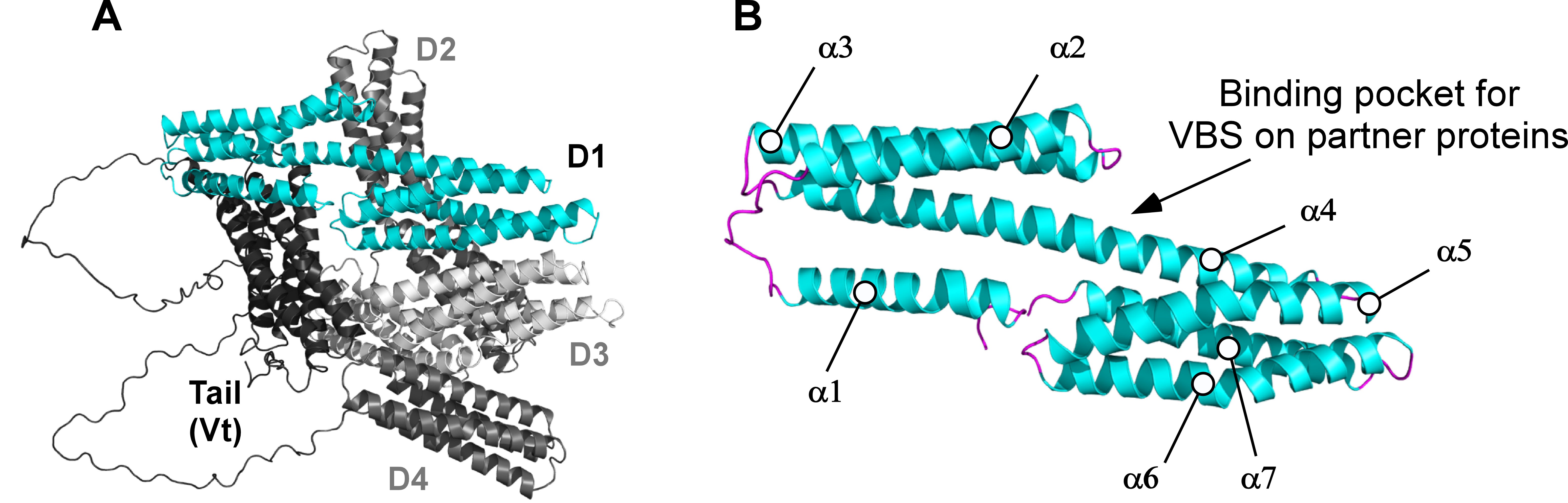
Figure 1. Crystal structure of vinculin full length and of vinculin D1 (vinculin head). A) Full-length vinculin model obtained from Alpha Fold database [28,29]. The vinculin domain 1 (D1) (amino acids 1 to 252), in the head of the molecule, is highlighted in cyan, and all the other domains (D2, D3, D4 and vinculin tail-Vt) are depicted in grey scale. B) Structure of vinculin D1 domain with its seven-α helices. α1, α2, α3, and α4 form the four-α helices bundle for classical VBS interaction.
Shigella are Gram-negative, non-motile, facultative anaerobic bacteria that infect the epithelium lining the terminal ileum, colon, and rectum in the gastrointestinal tract. Shigella is transmitted through feco-oral contamination from human-human transmission. These enteropathogenic bacteria are classified into four species (Shigella dysenteriae, Shigella flexneri, Shigella boydii, and Shigella sonnei) with multiple serotypes each [35].
Shigella´s invasion and epithelium disruption are mediated by an intricate and efficient coordination of various molecular machineries[32]. Although related to enteroinvasive E. coli (Escherichia coli), Shigella are non-motile, and they do not possess a flagellum. Shigella does not express any adhesin or curlin that would allow constitutive cell-binding activity. These pathogens use a specialized Type III Secretion Apparatus (T3SA), a macromolecular needle-like structure which it is capable to introduce a large number of effector proteins (including Ipa) to facilitate the invasion, replication, and dissemination [32]. Specifically, Shigella uses Ipa to leverage the host innate cellular processes such as actin dynamics and adhesive complexes to achieve early invasion and dissemination [36, 37]. In its inactive state, T3SA is capped by the tip complex composed of IpaB and IpaD invasins[38]. Upon contact with the target cell, IpaB and IpaC insert into the host cell membrane to form the translocation complex and transport type III invasion effectors across the host membrane. Entry of the IpaC in the cell cytosol induces the recruitment and activation of the Src tyrosine kinase, actin polymerization and consequent membrane ruffling. Furthermore, invasins IpgB1 and IpgB2 may target independently but synergistically Rac and Rho GTPases to promote efficient actin polymerization [39]. While Rac activation is implicated with Arp2/3 dependent polymerization of branched actin at membrane protrusions, Rho is associated with acto-myosin contractility [40]. Furthermore, activation of Rac GTPase antagonizes Rho, and viceversa [41] and their spatiotemporal coordination is crucial to regulate membrane protrusion dynamics [42]. This indicates that IpgB1 and IpgB2 specific activation of Rac or Rho regulate actin polymerization and cellular tension to promote invasion. Interestingly, analysis of mutants and their preferential localization at the cell-cell junction suggests that IpgB1 and IpgB2 may directly or indirectly target junctional structures [43, 44]. In addition to IpgB1 and IpgB2, IcsA, IpgD and IpaA are known to compromise the actin cytoskeleton directly or indirectly. For instance, the surface protein called IcsA (VirG) polymerizes actin to generate a flow that allows intracellular bacterial motility [45], which is essential for e.g., cell-to-cell spreading [46]. IpgD disassembles cortical actin by mediating hydrolysis of PI(4, 5)P2 into Pi(5)P, causing local detachment of the actin cytoskeleton from the plasma membrane to induce membrane protrusions at the site of bacteria invasion [47]. Finally, it has been recently discovered that IpaA interacts with vinculin and thus compromise the link between actin cytoskeleton and adhesion complexes of the host cell. Importantly, this may lead to defective mechanical homeostasis of the tissues with consequent loss of barrier and transport functions, due to loosen adhesions [48]. This loss of tissue integrity and function may result in a ¨leaky¨ epithelium with consequent watery and, in the worst cases, bloody diarrhoea, and in turn it compromises the host's immune system and overall patient´s health conditions [49]. Thus, while it has been formally categorized as an invasin, it has also been suggested that IpaA may play multiple roles during bacterial infections not only relegated to the invasion process [45]. As IpaA´s interference with cell mechanics and tissue homeostasis is central to the pathophysiology of Shigella infection, it is important to understand the molecular details of IpaA/vinculin interaction.
Among all invasins, IpaA has recently gained a lot of attention due to its ability to interact with vinculin, a major cellular hub crucial for tissue mechanotrasduction [50]. IpaA possesses three vinculin binding sites (VBS1 - 612 to 630 residues, VBS2 - 566 to 584 residues, and VBS3 - 492 to 510 residues) located at the C-terminus of the protein where they fold into the typical α-helix structure of all VBSs [51](Figure 2A). Similarities in structure and conserved residues (Figure 2B) between IpaA-VBSs and those of native proteins of FAs and AJs are suggestive of bacterial molecular evolution that led IpaA to mimic one of the most crucial modules essential to regulate epithelial tissue mechanics and physiology. It has been indeed suggested that Shigella uses ¨molecular mimicry¨ to hijack vinculin´s mechanical function to invade the target cell [7, 50]. Upon its injection in the host cell, IpaA preferentially interacts with the free cytosolic pool of vinculin (i.e., not engaged at FAs nor at AJs), which is recruited at the site of bacterial entry where they form adhesive structure between bacterium and host cell integrated with the bacterial-induced actin foci [7].
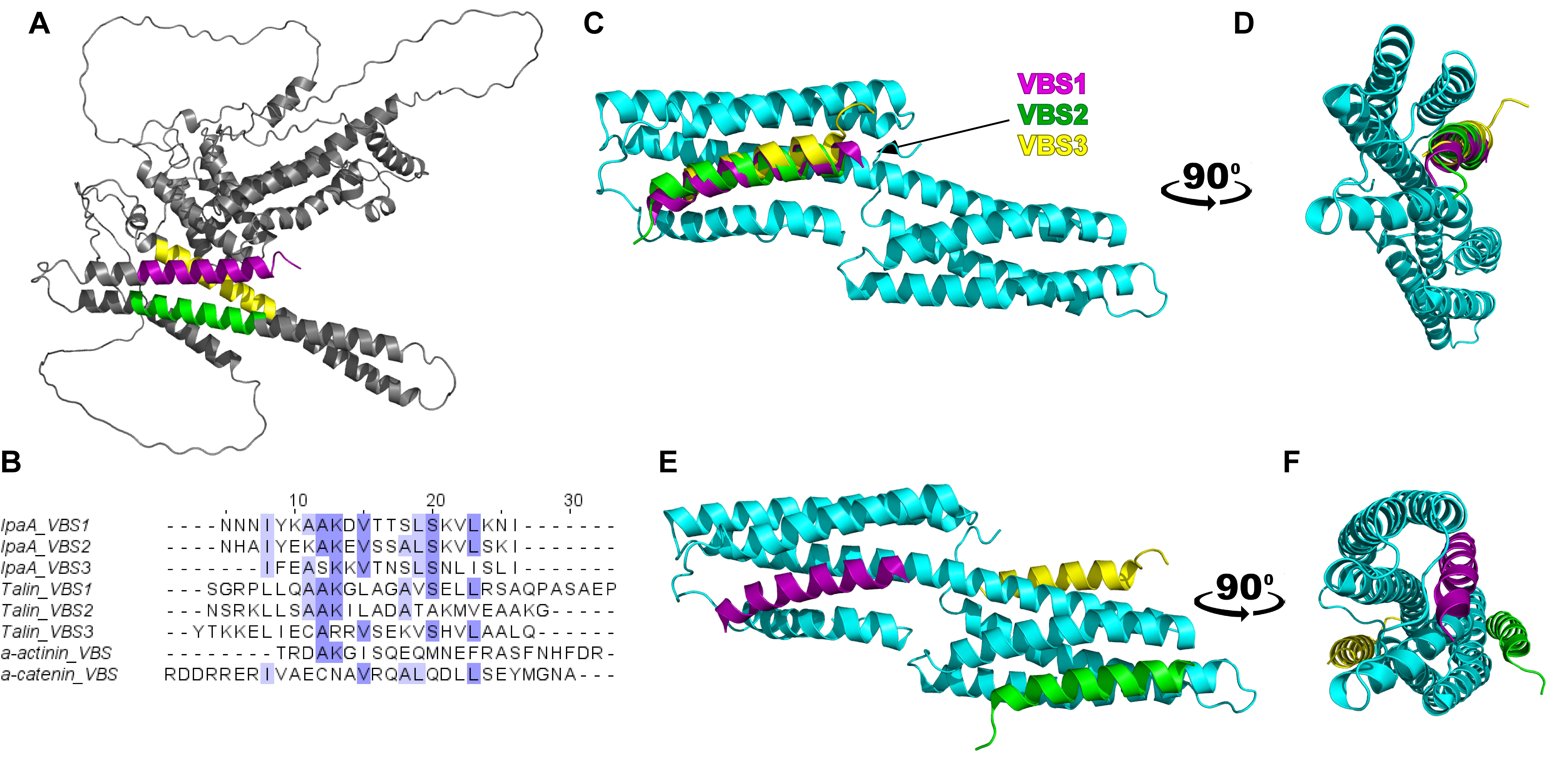
Figure 2. Proposed model for alternative Vinculin D1 / IpaA interactions. In all panels, vinculin D1 domain is depicted in cyan and IpaA vinculin binding sites VBS1, VBS2 and VBS3 are depicted in purple, green and yellow, respectively. A) Crystal structure of IpaA full length. B) Sequence alignment for vinculin binding sites from IpaA (IpaA_VBS 1, 2 and 3) [52], talin (Talin_VBS1,2 and 3) [53], α-actinin (α-actinin_VBS) [54] and α-catenin (VBS_ α-catenin) [55] shows a large level of homology between all VBS motifs. Alignment obtained using Clustal Omega at EMBL-EBI [56,57]. Conserved residues are highlighted in shades of blue in all six VBSs. C) Proposed structure model of interaction between human vinculin D1 with IpaA VBSs as isolated peptides. The three VBSs appear to overlap in their mode of interaction in the four-α helices pocket. D) Lateral view of C) with 90° rotation. E) Alternative proposed structure model of interaction between human vinculin D1 with IpaA VBS123 when considering static hindrance and site occupancy within the whole C-terminus segment (residues 483 - 633). The three VBSs are located at different sites as compared to C). F) Lateral view of E) with a 90° rotation. Models have been obtained using PyMOL2 software (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC., n.d.).
Like the modus operandi previously described for α-catenin and talin, the interaction between vinculin and VBSs of IpaA is thought to induce a key conformational change. This change affects the head-tail interaction of vinculin and forces the protein into its most mechanically open and active state where it can bind to F-actin [44]. It has been shown that all three IpaA-VBSs as isolated peptides can interact with the 4-helix pocket (5-helix interaction mechanism) of the mechanosensitive domain of vinculin (vinculin D1, Figure 2C- D)[7, 51]. However, it is not clear how the three VBSs interact with vinculin when interconnected by the linkers present in the IpaA full structure. Recently, it has been reported [59] that the three interconnected VBSs (residues 483 - 633) can bind to a single vinculin in different locations (Figure 2E-F) with VBS1 locating in the 4 α-helices pocket in D1, VBS2 binding to α5 and α6 of D1 and VBS3 inserting into the D1D2 cleft region [59]. This last interaction could induce a rotation of D2 respect to D1, thus loosening vinculin structure and provoking the release of the tail to activate actin binding to vinculin. In addition to these recent discoveries, it must be noted that structural and functional studies of vinculin/IpaA interaction using full-length proteins are still lacking. When considering the whole IpaA molecule (Figure 2A), VBS3 is clearly located in a hidden position, and it is connected through a flexible linker to the IpaA bulky N-terminus portion. Interestingly, such steric hindrance is also observed in various mechanosensitive vinculin-binding partners (i.e., α-catenin and talin) that can interact with vinculin when cellular forces (e.g., actin contractility) cause force-dependent conformational changes [60–62]. While this is only speculative, a similar mechanism may be required to mediate IpaA/ vinculin interaction and further investigation would be needed.
Other interesting venues of further exploration concern the mechanical roles of IpaA during invasion and as virulence factor aiding spreading of the disease. The actual mechanisms by which IpaA functions as invasin is still unclear and debated. It has been recently proposed that all three VBSs bind to different vinculins at site of Shigella entry to promote vinculin oligomerization with consequent formation of actin bundles that may stabilize adhesion between Shigella and the host cell [63] (Figure 3). On the other hand, IpaA/vinculin interaction may dysregulate cell mechanics in a number of ways. For instance, IpaA may compete with α-catenin and talin and sequester/deplete the pool of cytosolic vinculin available to regulate cell adhesions dynamics. In addition, it has been reported that IpaA can localize at the FAs where it could compromise cell-substrate interaction by interfering with vinculin connection with its partners [7]. At this site, it could also synergistically act with other virulence factors, such as IpgB1 and IpgB2, that have been reported to accumulate at adhesion sites to control actin polymerization and depolymerization [43, 44]. Finally, it has also been shown that IpaA-VBS3 can interact with talin leading to filopodial adhesions and efficient capture of Shigella [63, 64]. It has been proposed that stable filopodia are required to facilitate adhesion of the bacteria with the cells[51, 65]. However, it is also conceivable that this same can compromise normal formation of protrusive structures in the host cells, such as filopodia and lamellipodia, needed for cell migration during e.g., wound healing. Thus, it is not too farfetched to hypothesize that, in addition to functioning as an invasin, IpaA may play a key role in disrupting the normal mechanical regulation of the epithelial tissue by sequestering cytosolic vinculin, by impairing vinculin interaction with its binding partners at FAs and AJs, and by compromising actin structures and dynamics. These deregulations may compromise epithelial tissue homeostasis and fluid balance, thus inducing diarrhoea, contamination of water and surfaces, and spread of the bacterium. Finally, it should also be considered that IpaA could affect cell-cell adhesion with consequent loss of epithelial barrier function. Importantly, this may allow Shigella to directly access the basolateral side of the cells, which is the preferred site of bacterial entry [45, 66] and thus it would promote further invasions. In extreme cases, disruption of the barrier function would further allow all sort of opportunistic bacteria and viruses to access blood stream and lymphatic system, and it may thus initiate septic response in patients [67].
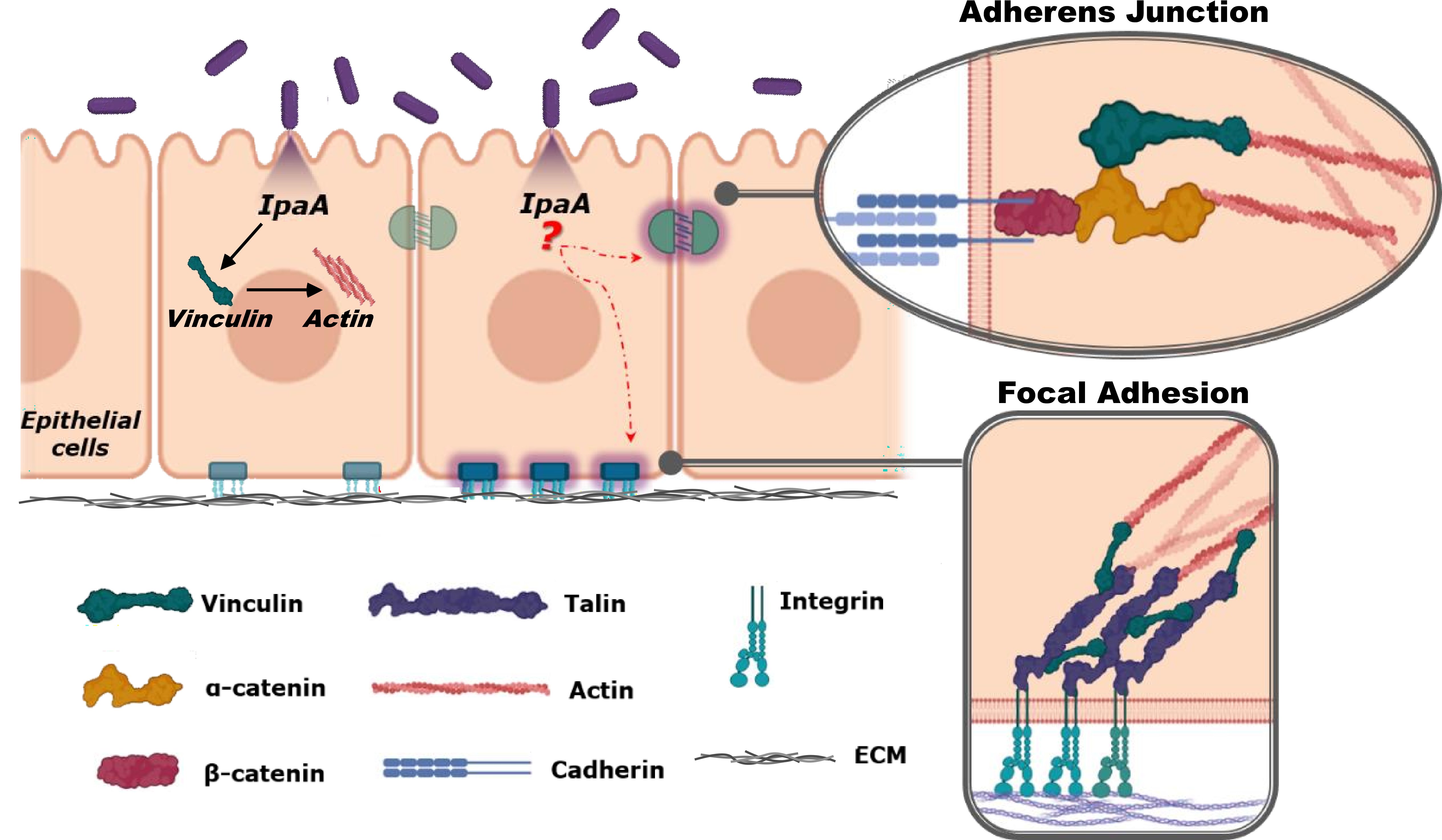
Figure 3. Shigella creates its own adhesive contacts and may interfere with the adhesion’s complexes of epithelial cells. Schematic representation of a simple epithelial tissue depicting mechanically regulated adhesive complexes (i.e., FAs and AJs). Upon injection of IpaA into epithelial cells cytosolic vinculin is recruited together with actin to facilitate bacterial invasion. Possible targeting of adhesive complexes and consequences in terms of regulation of cell and tissue mechanics have not been determined. Similarly, details of the molecular mechanism of IpaA-vinculin interaction and consequences for cellular and tissue mechanics are still not clear. Image was created with Biorender.
The recent COVID-related social and medical emergency has dramatically exposed the susceptibility of our global society to infection diseases. Undoubtedly, it has also demonstrated the need to prepare for such eventualities by building an in-depth understanding of the potential targets for therapeutic intervention. In this sense, emergence of drug resistant strains of Shigella is reason for great concerns [68] and, thus, it is of vital importance to better understand the molecular mechanisms that lead to infection and the worst symptomatic outcomes of the disease. Due to its specific interaction with vinculin, the invasin IpaA could be a very important virulence factor to monitor. This interaction is used by the bacterium to mechanically power its ingress into the host by parasitically controlling the cell adhesion complexes and actin active processes. In this review, we have also highlighted that likely outcome, either as a by-product or intended effect of the infection, of IpaA/vinculin interaction is mismanagement of cellular and tissue process in charge of maintaining mechanical homeostasis of the epithelium. However, direct evidence and systematic analysis at the cell and tissue level are scarce. Thus, we believe that future research should address how IpaA/vinculin mechanomolecular interaction affects cells and tissues and, at the system level, its significance for epithelial barrier function, water balance, transport, and metabolism. This would allow for the understanding of the connection between the mechanistic details of the infection and the insurgence of the pathology.
On the other hand, a deeper understanding of IpaA/vinculin interaction in full-length proteins would be important to pave the way for directed design of interfering molecules. For instance, while the three VBSs of IpaA have quite a large homology with VBSs of native proteins, having the full picture of the protein-protein interfaces involved would clarify if there is the possibility to specifically compete with vinculin/IpaA interaction without compromising other physiological interactions between vinculin and native proteins. A similar point could be raised regarding the IpaA head, i.e., the large portion of the protein that does not contain the three VBSs, whose functions is still largely unclear.
In conclusion, we believe that adopting mechanical and multiscale perspectives to study the molecular details of Shigella invasion and of pathogen-host interaction could pave the way for the development of entirely novel and much needed therapeutic interventions.
The authors gratefully acknowledge funding support from ANID/SCIA/ACT192015, ANID FONDECYT Regular 1210872, ANID FONDEQUIP MEDIANO EMQ210101 and EQM210020, and support by seed fundings from the Pontificia Universidad Catolica de Chile (Puente -2022-13).
Author contributions
All authors discussed and wrote the manuscript.
The authors have no conflicts of interest to declare.