Original Article - DOI:10.33594/000000634
Accepted 11 April, 2023 - Published online 24 June, 2023
1Department of Gastroenterology, Hannover Medical School, Germany;
2Department of Gastroenterol- ogy, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Background/Aims: Mice deficient for the canalicular phospholipid transporter MDR2 (ABCB4) develop sclerosing cholangitis due to high biliary concentrations of monomeric bile acids. This study determines whether a selective reduction in biliary bicarbonate output, secondary to the deletion of the hepatocyte-expressed carbonic anhydrase CAXIV (Car14) aggravates the bile acid-induced damage observed in the mdr2-/- mouse model. Methods: Bile flow was measured gravimetrically and HCO3- output by microtitration before and during stimulation with intravenously applied tauroursodesoxycholic acid (TUDCA) in car14-/-mdr2-/- (abcb4-/-), car14-/-/mdr2+/+, car14+/+/mdr2-/- and wt mice. Cholangiocyte proliferation, hepatic inflammation and fibrosis was studied by gene and/or protein expression for proinflammatory and profibrotic cytokines, cholangiocyte proliferation markers, and by (immuno) histochemical assessment. The impact of Car14 deficiency was also assessed in a xenobiotic cholangitis model. Results: TUDCA stimulated HCO3-output was significantly increased in 6 week old mdr2-/- mice, and significantly decreased in both car14-/- as well as car14-/-/mdr2-/-mice, compared to wt, while bile flow was unaltered. Both bile flow and HCO3- output were significantly decreased in 11 week old mdr2-/-, and more so in car14-/-/mdr2-/- mice. Loss of Car14 significantly increased inflammatory liver injury and cholangiocyte proliferation, and aggravated liver fibrosis in car14-/-/mdr2-/- mice compared to mdr2-/- mice. In contrast, the absence of Car14 did not affect the hepatic functional and morphological alterations in 3,5-diethoxycarbonyl-1,4-dihydroxychollidine (DDC) fed mice. Conclusions: Car14 deletion reduced biliary HCO3- output and aggravated the functional, inflammatory and morphological alterations in the liver of mdr2-/-mice. These results demonstrate the importance of sufficient hepatocellular bicarbonate output in the protection of the hepatobiliary epithelium against toxic bile acids.
The biliary ducts are exposed to very high luminal bile acid concentrations, which are toxic to other epithelia [1]. The biliary epithelium has been suggested to be protected against bile acid induced cytotoxicity by a locally generated “biliary HCO3- umbrella”, whereby an alkaline microclimate in the glycocalyx of the cholangiocyte, generated by receptor mediated cholangiocyte HCO3- secretion, neutralizes acidic moieties such as monomeric bile acids [2]. Support for this hypothesis is provided by several findings in human cholestasis diseases. For example, a downregulation of the anion exchanger AE2 (SLC4A2), which is considered essential for cholangiocyte HCO3- output [3], in primary biliary cholangitis (PBC) by epigenetic mechanisms is considered central to the pathogenesis of the disease [4, 5]. In the fibrosing cholangiopathy of cystic fibrosis (CF) associated liver disease, the cholangiocyte Cl-/HCO3- channel CFTR is defective [6].
However, hepatic bile as well as HCO3- is predominantly derived from hepatocellular secretion [7]. Early work in isolated perfused livers and isolated hepatocytes studied the interrelationship between bile acid dependent and independent bile flow and HCO3- output [8, 9]. In parallel, the hepatocyte canalicular membrane transporters for HCO3- were studied functionally [10, 11] and immunohistochemically [12]. The CFTR anion channel has been localized in cholangiocytes but not hepatocytes [13]. It was found that while the secretin stimulated fluid and HCO3- secretion was CFTR dependent and originated in the bile ducts, bile salt stimulated fluid and HCO3- secretion was predominantly of hepatocellular secretion and was CFTR independent [14].
Hydrophilic bile salts stimulate an increase in hepatocellular fluid and bicarbonate secretion, and their protective role in experimental cholestatic liver disease is undisputed [15-17]. But how important is the hepatocyte bicarbonate output, independent of the fluid secretory response?
The recent discovery of a strong expression of carbonic anhydrase 14 (CAXIV, Car14) in hepatocytes and the generation of a car14-/-mouse model may provide a suitable animal model to study this question. Carbonic anhydrases (CAs) are a family of functionally related proteins that catalyze the reaction H+ + HCO3- ↔ CO2 + H2O [18-20]. Both cytoplasmic and membrane bound CAs are important for the supply of protons or base to acid/base transporters, and may increase their transport rates several fold if existing in a “HCO3- transport metabolome” with the respective transporter [21-23]. In a recent study, young and adult Car14 deficient mice displayed reduced biliary HCO3- output without a reduction in bile flow [24]. Despite a significant decrease in hepatic HCO3- output into bile, pathological changes in the bile ducts developed only in aged mice, and were very mild [24]. In addition, the present study revealed no significant alterations in the expression of the major hepatocyte and cholangiocyte acid/base and bile salt transporters between car14-/- and wt mice. A potential reason for the relatively minor protective function of HCO3- in this model may be that the murine bile acid pool, which consist predominantly of hydrophilic bile acids, does not have the same damaging potential as the hydrophobic bile acids, which are more predominant in human than murine bile [25].
Therefore, we searched for mouse models with a more toxic bile acid pool, in order to cross them with the car14-/- mice. In the mdr2-/-(abcb4-/-) mouse model of primary sclerosing cholangitis, bile duct injury develops as a consequence of defective biliary phospholipid (PL) secretion and subsequent increase of free non-micellar and potentially toxic bile acid (BA) concentrations in bile [26-28]. This model seemed ideal to us to explore a selective role of HCO3-, independent of bile flow, in the protection of the intrahepatic bile ducts against toxic bile acids. We therefore evaluated the effect of Car14 ablation on bile flow, biliary HCO3- output, and the severity of liver damage in male and female mdr2-/-(abcb4-/-) mice in juvency and young adulthood. As a control model, we employed a 3, 5-diethoxycarbonyl-1, 4-dihydroxychollidine (DDC) feeding, in which a protective role of biliary HCO3- is not anticipated (but cannot be ruled out) [29-31].
Animal models of the studyAll mice were bred on the C57/B6 background in the animal facility of Hannover Medical School. Mice were maintained with controlled light/dark cycles and free access to water and food. All experiments were approved by the Local Institutional Animal Care and Research Advisory Committee at the Hannover Medical School and authorized by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES) (TVA Nr. 33-12-42502-04-15-1847). The experimental procedures performed and the type of anaesthesia used were according to university and national guidelines and are explained below. The car14-/- (B6.129S1-Car14tm1sly) mice were originally created in the group of William Sly in the Edward A. Doisy Department of Biochemistry and Molecular Biology [32] and was provided by Prof. Gerolf Gros, Institute of Physiology, MHH. The mdr2-/-(FVB.129P2-Abcb4tm1Bor, six times backcrossed on C57/B6) mice were provided by Prof. Arndt Vogel, MHH. Car14 deficiency was generated in the mdr2-/- mouse strain by breeding with heterozygotes. 4 genotypes were obtained: car14+/+/mdr2+/+, car14-/-/mdr2+/+, car14+/+/mdr2-/-and car14-/-/mdr2-/-. In each set of experiments, littermates were used for the different genotypes, and a similar number of male and female mice were used.
To evaluate the possibility that the loss of Car14 exerts detrimental or protective effects to injury unrelated to hydrophobic bile acid injury, we also studied the DDC feeding model of sclerosing cholangiopathy. Administration of DDC induces an increase in the secretion of hepatotoxic protoporphyrins, concomitant with formation of protoporphyrin plugs. The latter induces an obstruction in the small bile ducts, which initiates cholestasis, leading to sclerosing cholangitis and pronounced biliary fibrosis accompanied by ductular proliferation. 8-10 week old mice car14+/+ and car14-/- mice were fed with 0.1% DDC supplemented diet for a period of 3 weeks [33]. DDC supplemented diet was purchased from Altromin Spezialfutter GmbH, Germany. Age matched mice fed with isocaloric diet were used as a control group of all experiments. After beginning with the DDC diet, the mice were weighed every three days. Initial weight loss was due to avoidance of the chow, then stabilized and was followed by a weight gain, but from 10-14 days there was progressive weight loss. The mice, which lost more than 30% from their initial body weights, were sacrificed and excluded from this study. Feeds and bedding were weighed and changed twice per week to provide an estimate of food intake.
ReagentsTUDCA (Tauroursodeoxycholic acid) (Calbiochem/MerckBiosciences), Mayer's hematoxylin, Eosin Y solution, Picric acid, Direct Red 80, 30% H2O2, EDTA (Sigma Aldrich), Xylene (J.T. Baker GmbH), Permount, Fast Green FCF (Thermo Fisher GmbH) , Goat Serum (Vector Laboratories), anti-CK19 antibody (DSBH of Iowa University) , AEC (red) substrate Kit, phosphate buffered saline, goat anti-mouse IgG (Life Technologies GmbH). All other chemicals were obtained from Applichem GmbH, Germany, unless mentioned otherwise.
In vivo biliary secretion experiments
Mice were fasted for a minimum of 4 hours before the initiation of the biliary drainage operation. The in vivo biliary drainage was performed as previously described [24]. In brief, isoflurane (Forene; Abbott Germany, Wiesbaden, Germany) anesthetized, tracheally intubated and mechanically ventilated (MiniVent Type 845; Hugo Sachs Electronik, March-Hugstetten, Germany) mice were equipped with a rectal thermistor probe to maintain the body temperature at approx. 37.5 °C. A left carotid artery catheter was used to monitor blood pressure (PowerLab system, AD Instruments, Hastings, UK) and for infusing an alkaline solution at a rate of 0.1ml/h to correct the systemic acid–base balance at the following composition: 200mM Na+, 100mM CO32-, 5mM K+ and 5mM Cl−, as previously described [34, 35]. A left jugular vein catheter was used to infuse 0.3 ml/h PBS, with or without tauroursodeoxycholate (TUDCA) infusion at the rate 600 nmol TUDCA/min [36, 37]. The abdomen was opened, the gallbladder neck ligated, and the common bile duct was cannulated with a polyethylene tubing made very thin at the tip over a flame. The abdominal cavity was closed and an approx. 20 min equilibration period ensued before the start of the experiment. Baseline values were collected for 40 min, after which the mice were infused with TUDCA via carotid artery for another 60 min. Mice were killed by cervical dislocation at the end of the collection period. Livers were excised, weighed, dissected and processed for histology and laboratory examinations.
Blood pressure was continuously monitored and if too low, either the isoflurane concentration was adjusted, or the infusion speed was increased, or both. Blood samples were taken for blood gas analysis at the end of the stabilization period, and adjustments were made by increasing or decreasing the infusion speed of the NaCO2 solution. Another sample was taken at the end of the experiment for blood gas and hematocrit analysis. The sample for blood gas was taken from the carotid artery into a heparinized glass capillary (Clinitubes, Radiometer, Copenhagen, Denmark), and immediately analyzed in a Radiometer blood gas analyzer (ABL-5 Blood Gas Analyzer, Radiometer, Copenhagen, Denmark) immediately before coagulation. The sample for hematocrit was taken into another glass capillary and spun in a Hettich Hematocrit 210 hemocytometer (Andreas Hettich GmbH, Tuttlingen, Germany).
Bile flow was measured gravimetrically as previously described [24]. The rate of luminal alkalinization was determined as described [24] via back titration of the bile sample to pH 4.5 with 5mM HCl under continuous N2 gassing using pH-stat equipment (PHM82 Standard pH meter, Radiometer, Copenhagen, Denmark [38, 39]. The rates of bile alkalinization are expressed as micromoles of the base secreted per hour (µmol/h).
Liver tissue examination
We followed the guidelines for standardized work-up for mouse models of the International PSC Study Group (IPSCSG) [27, 28]. A detailed figure of how the liver was cut is found in the cited publication, however, we chose to use whole liver lobes for gene expression studies, not to bring in bias to the experiments. Liver tissue was collected immediately after sacrifice. Tissue fixation and processing, as well as the histological and immunohistochemical analyses were performed as previously described [24].
qPCR protocol
RNA extraction and qPCR was performed exactly as previously described [24] in a Rotor Gene Q device. The primers are given in Supplementary Table 1. For all gene expression data, the y- axes denote the 2 ^ -deltaCt values.
Western analysis
Mouse liver tissue was homogenized in RIPA lysis buffer (25 mM Tris, 150 mM NaCl, 1 % Nonidet P40, 0.5 % Sodium deoxycholate, 0.1 % SDS, pH 7.5) supplemented with a cocktail of protease inhibitors (40 µg/ml PMSF, 20µg/ml Leupeptin, 20µg/ml Pepstatin A, 20µg/ml Antipain, 4 mM Benzamidin, 1 mM DTT) using an Ultra‑Turrax homogenizer (IKA) set for 3 times each for 10 secs at speed 4, with 10 sec intervals on ice. The homogenate was further cold treated by 10 up and down strokes with a Potter homogenizer at 1000 rpm (Braun Biotech International) and the debris was sedimented by centrifugation at 10000 rcf at 4 °C for 10 min. The protein content of the lysate was determined by the Bradford method (BioRad). 30 µg total protein was resolved by 10 % SDS-PAGE (Mini PROTEAN Tera Cell system; BIO-RAD) and wet transferred to PVDF membrane (Mini Trans-Blot Cell system; BIO-RAD) at 300 mA for 120 min. The membrane was then blocked in the TBS containing 0.1 % Tween 20 and 5 % non-fat milk (BIO-RAD). Primary antibodies against Cytokeratin 19 (clone TROMA-3; DSHB) at 1:5000 dilution or Vinculin (V9131, Sigma-Aldrich) at 1:10000 dilutions were prepared in blocking buffer and exposed to the membrane overnight at 4 °C. After proper washing steps before and after incubating the membrane with HRP-goat anti-mouse secondary antibody (Thermo Scientific) at 1:10000 dilution for 40 min at room temperature, the corresponding protein signals were visualized by ECL Western Blotting Detection Reagents (GE healthcare#RPN2209) and developed on an X-ray film. The bands were scanned and quantified using ImageJ software.
Statistical analysis
Statistical analyses were performed using the Prism analysis program (Graphpad 8.0, San Diego, CA, USA). The statistical significance of data was tested via repeated measures analysis of variance. To test differences within a group, one-way ANOVA was used followed by a Tukey post-hoc test. Between groups, two-way ANOVA was used followed by a Bonferroni post-hoc test. A p-value of less than 0.05 was considered significant. *p <0.05, **p<0.005, ***p≤0.0001. Comparisons of two groups which passed the Shapiro-Wilke normality test were compared with the two-tailed Student t-test. All data are presented as means ± standard error of the means (SEM).
Car14 mRNA expression in the livers of mdr2-/- and wt littermates.To test a potentially protective effect of Car14 expression against toxic bile acids in vivo, we hypothesized that deleting Car14 in a murine cholestatic model is only a meaningful approach if a robust expression of Car14 in the inflamed liver is evident. Car14 mRNA expression was therefore determined in 6 and 11 week old mdr2-/- and wt littermate livers (Fig. 1). Car14 mRNA expression was slightly higher in mdr2-/- compared to wt livers at 6 weeks of age and similar at 11 weeks. Therefore, the expression levels of Car14 are high enough in the inflamed livers to test the relevance of its genetic deletion on the pathologic phenotype of the mdr2-/- liver.
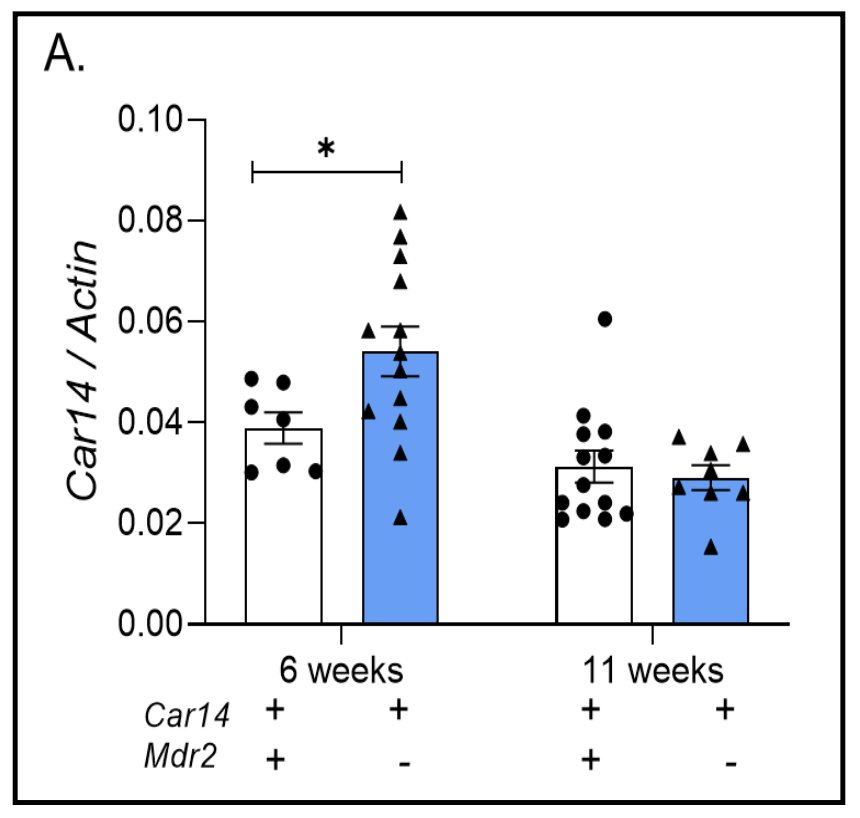
Figure 1. Carbonic anhydrase 14 expression in wt and mdr2-/- liver. (A) Robust Car14 mRNA expression was observed in wt and mdr2-/- liver tissue, with significantly higher Car14 expression in young (6 weeks/age) mdr2-/- than wt liver. No difference seen at 11 weeks, likely due to the progressive disease in the mdr2-/- liver, in addition to an age-related effect. Each dot represents the liver tissue of one mouse.
Loss of Car14 leads to a reduction in bile flow and strongly interferes with biliary HCO3- output in mdr2-/- livers.Car14 deficiency was generated in the mdr2-/- mouse strain by breeding with heterozygotes. 4 genotypes were studied:car14+/+/mdr2+/+, car14-/-/mdr2+/+, car14+/+/mdr2-/-, car14-/-/mdr2-/-. Biliary fluid and HCO3- secretion were determined in 6 week and 11 week old mice. In 6 week old mice, basal bile flow rates were not significantly different between the 4 genotypes, but TUDCA stimulated bile flow was significantly reduced in car14-/-/mdr2-/- mice (Fig. 2A). In contrast, biliary TUDCA stimulated HCO3- output was increased in the car14+/+/mdr2-/- compared to wt mice, but was decreased in car14-/-/mdr2+/+, and to a significantly greater extend in car14-/-/mdr2-/-mice (Fig. 2B). In 11 week old mice, TUDCA stimulated bile flow rates were not significantly different between car14+/+/mdr2+/+ and car14-/-/mdr2+/+ mice, but were significantly reduced in car14+/+/mdr2-/- and to an even higher degree in car14-/-/mdr2-/- mice (Fig. 2C). Biliary TUDCA stimulated HCO3- output was significantly decreased in car14+/+/mdr2-/-, car14-/-/mdr2+/+, and most strongly in car14-/-/mdr2-/- mice (Fig. 2D).
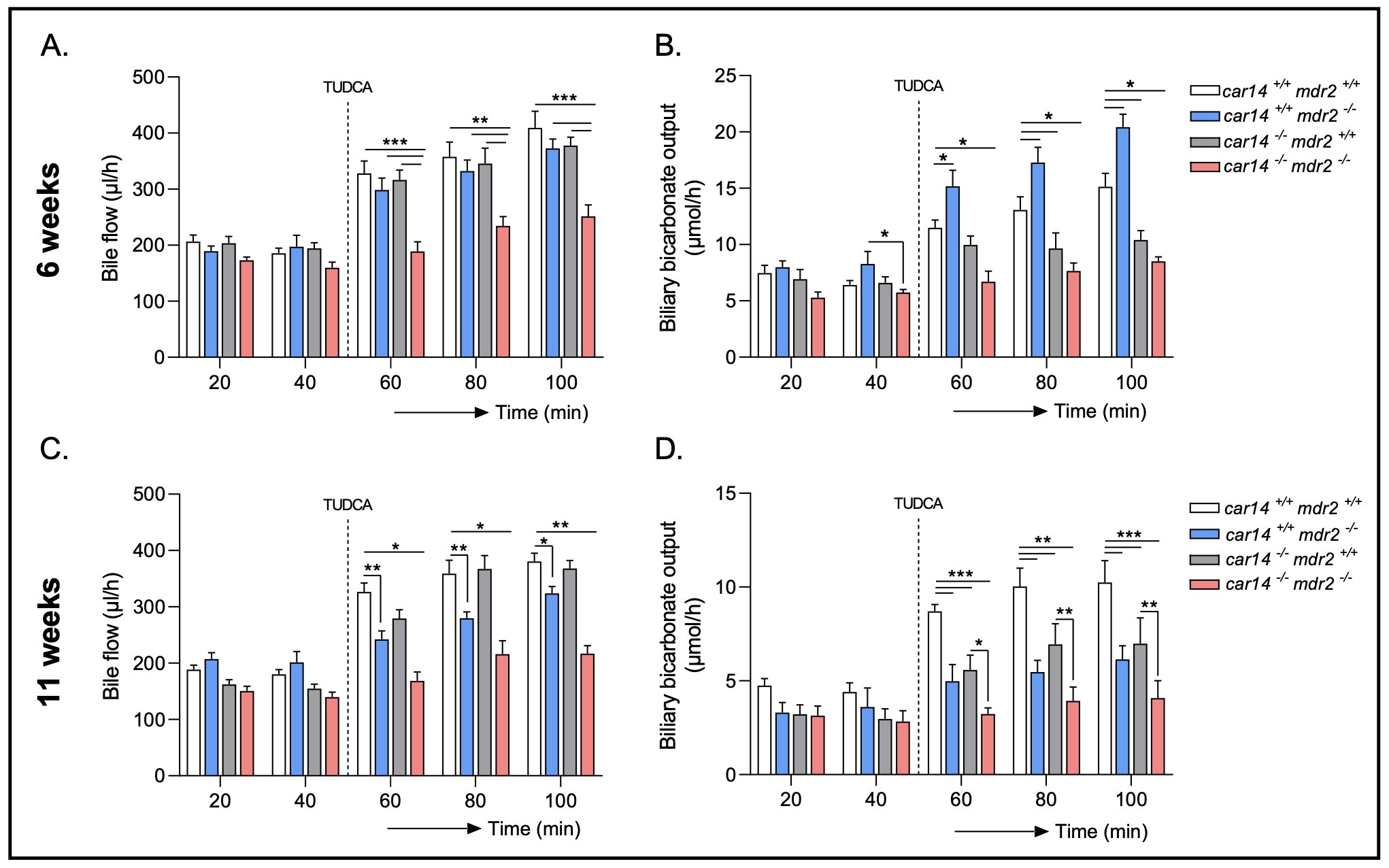
Figure 2. Bile flow and hepatobiliary HCO3- output in car14+/+/mdr2+/+, car14+/+/mdr2-/-, car14-/-/mdr2+/+ and car14-/-/mdr2-/- mice. (A, C) Bile flow rates in the basal state and after TUDCA stimulation in the four different genotypes. (A) At 6 weeks/age, no significant differences were observed in flow rates from car14+/+/mdr2+/+, car14-/-/mdr2+/+ and car14+/+/mdr2-/- common bile ducts before and during TUDCA stimulation (dissolved in PBS infused at the rate 600 nmol/min), while a significant decrease in TUDCA stimulated car14-/-/mdr2-/- bile flow rate. n= 6, 6, 7 and 8 for the 4 genotypes. (C) At 11 weeks/age, the bile flow rates were not different between car14+/+/mdr2+/+ and car14-/-/mdr2+/+, and did not differ from the flow rates of the younger mice with these genotypes. In contrast, the car14+/+/mdr2-/- mice had developed reduced TUDCA induced bile flow rates, and the car14-/-/mdr2-/- had further decreased TUDCA stimulated bile flow rates. n=8 for all genotypes. (B, D) Hepatobiliary HCO3- output rates in the basal state and after TUDCA stimulation in the four different genotypes. (B) At 6 weeks/age, car14+/+/mdr2-/- mice had significantly higher HCO3- output rates than car14+/+/mdr2+/+ mice, and both car14-/-/mdr2+/+ and car14-/-/mdr2-/- mice had significantly lower rates. n=6, 6, 7 and 8 for the 4 genotypes (D) At 11 weeks of age, car14+/+/mdr2-/-, car14-/-/mdr2+/+ and car14-/-/mdr2-/- mice displayed significantly lower TUDCA induced HCO3- output rates than car14+/+/mdr2+/+ mice, with the most dramatic relative reduction to week 6 in the car14+/+/mdr2-/- mice. n=8 for all genotypes. p values represent significant differences within the group of 4 genotypes, compared with two-way ANOVA followed by a Bonferroni post-hoc test.
Loss of Car14 aggravates liver injury in mdr2-/- livers.Histological analysis was performed in livers at 3, 6 and 11 weeks of age. Already at 3 weeks of age, H&E showed an increased periductal neutrophil infiltration in car14-/-/mdr2-/- livers (Fig. 3A, upper panels). At 6 weeks of age, the portal triad infiltration was conspicuous; the bile duct lumina were narrowed in car14+/+/mdr2-/- livers and more markedly so in car14-/-/mdr2-/- livers (Fig. 3A, middle panels). The inflammatory infiltrate became very prominent in the 11 week old livers, again with more severe pathologic alterations in car14-/-/mdr2-/- than in car14+/+/mdr2-/- livers (Fig. 3A, lower panels).
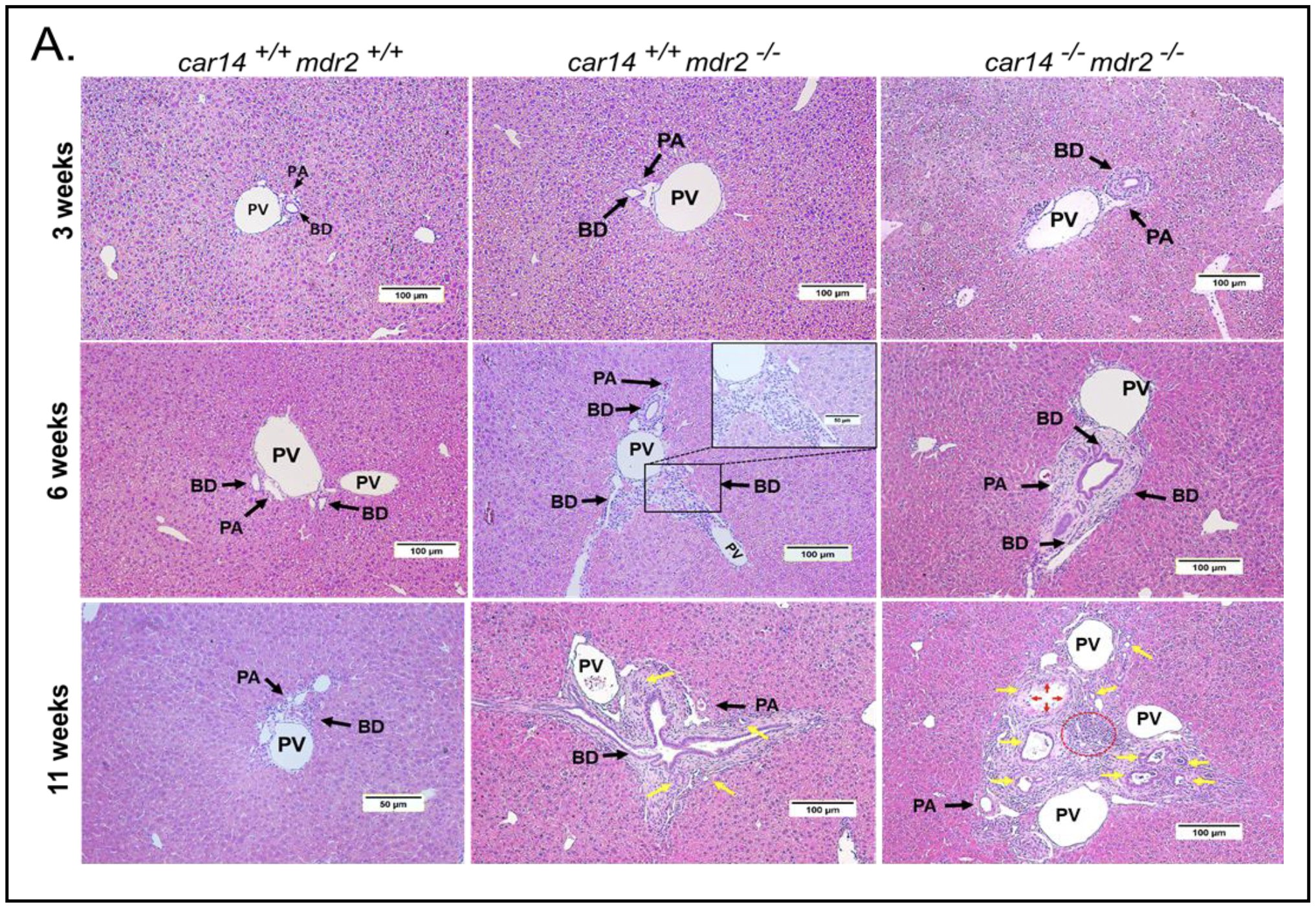
Figure 3A. Histopathological signs of liver damage in 3, 6, and 11 week old car14+/+/mdr2+/+, car14+/+/mdr2-/- car14-/-/mdr2+/+ and car14-/-/mdr2-/- mice. (A) Upper panel: H&E staining revealed no conspicuous abnormalities in the 3 week old car14+/+/mdr2-/- livers, but demonstrated mild bile duct wall thickening and neutrophil infiltration in car14-/-/mdr2-/- livers. Middle panel: Inflammatory infiltrates in periportal areas and bile duct thickening in 6 week old car14+/+/mdr2-/- and car14-/-/mdr2-/- livers. Lower panel: Severe periductal inflammatory changes in the portal triads of the car14+/+/mdr2-/- and car14-/-/mdr2-/- livers. Car14-/-/mdr2+/+ did not show histopathologic abnormalities in comparison to wt during the observed time span and are not shown BD: bile duct, PV: portal vein. PA: hepatic artery. Yellow arrows: Severe ductular proliferation; Red arrows: Atrophy and death of BECs and Red dotted line: Severe inflammation area.
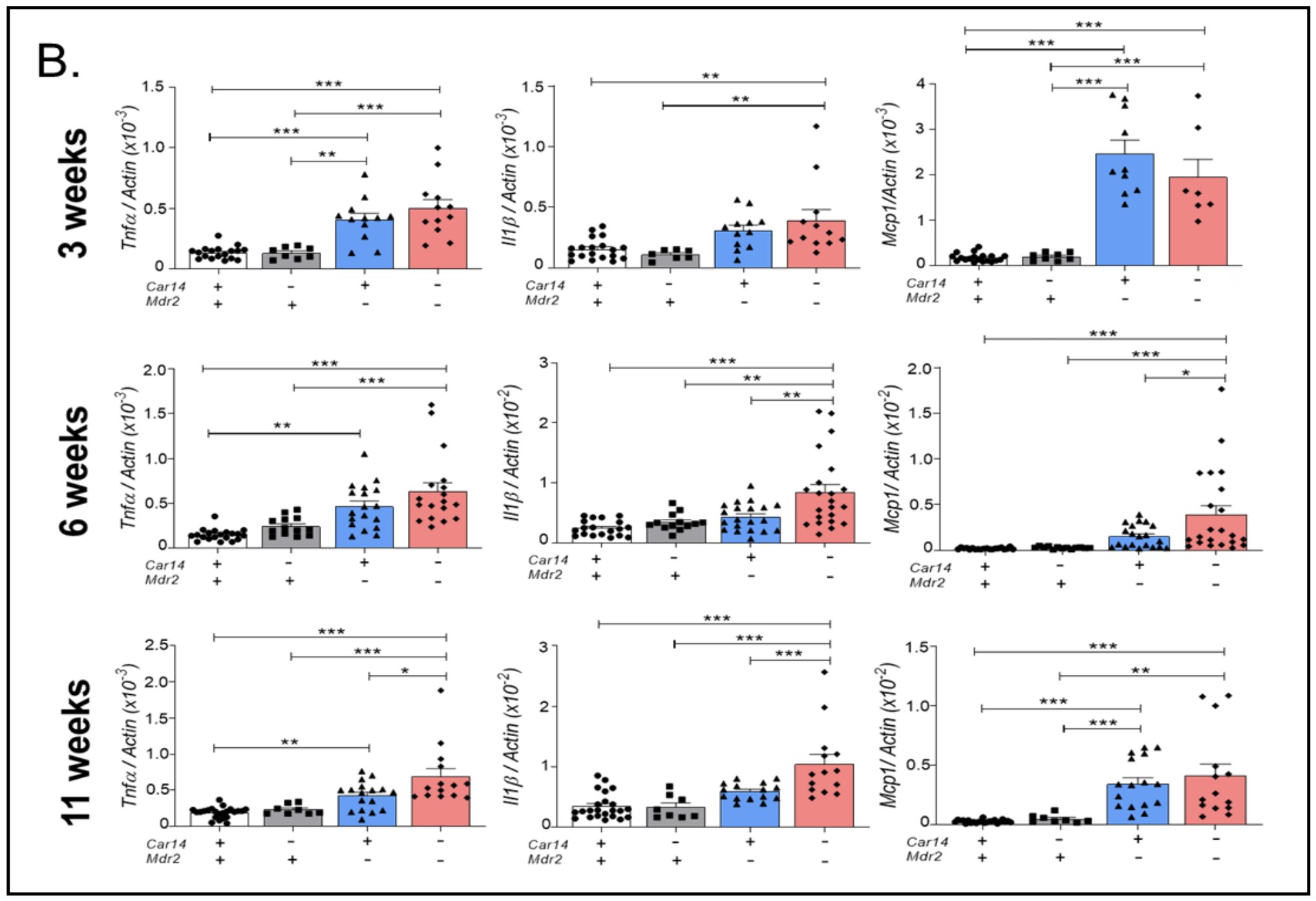
Figure 3B. Pro-inflammatory cytokine mRNA expression in 3, 6, and 11 week old car14+/+/mdr2+/+, car14+/+/mdr2-/- car14-/-/mdr2+/+ and car14-/-/mdr2-/- mice. (B) mRNA expression levels for proinflammatory cytokines Tnfα, Il1β, Mcp1 in the four genotypes. Upper panel: While the expression of all three proinflammatory cytokines were already significantly elevated in both car14+/+/mdr2-/- and car14-/-/mdr2-/- compared to car14+/+/mdr2+/+ and car14-/-/mdr2+/+ liver tissue, no difference was seen in the former two genotypes. Middle panel: At 6 weeks of age, mRNA expression levels for Tnfα, Il1β, Mcp1 were significantly elevated in both car14+/+/mdr2-/- and car14-/-/mdr2-/- compared to car14+/+/mdr2+/+ and car14-/-/mdr2+/+ liver tissue, and all except Tnfα were significantly higher in the car14-/-/mdr2-/- than the car14+/+/mdr2-/- livers. Lower panel: At 11 weeks of age, significantly higher mRNA expression levels for Tnfα, Il1β, Mcp1 in both car14+/+/mdr2-/- and car14-/-/mdr2-/- compared to car14+/+/mdr2+/+ and car14-/-/mdr2+/+ liver tissue, with Tnfα and Il1β expression being significantly higher in the car14-/-/mdr2-/- than the car14+/+/mdr2-/- livers , n number: Each dot represent a liver sample from one mouse. p values represent significant differences within the group of 4 genotypes, compared with two-way ANOVA followed by a Bonferroni post-hoc test.
mRNA expression of inflammatory markers in the car14+/+/mdr2+/+, car14-/-/mdr2+/+, car14+/+/mdr2-/- and car14-/-/mdr2-/- livers.mRNA expression of genes for inflammatory cytokines (Tnfα, Il1β, Mcp1) was assessed in the lobe 3 of the liver of 3, 6 and 11 week old mice (Fig. 3B). No significant difference was seen between car14+/+/mdr2+/+ and car14-/-/mdr2+/+ livers (both named control in the subsequent text). All the above named genes were already significantly elevated in car14-/-/mdr2-/- compared to controls at 3 weeks of age (Fig. 3B, upper panel), and all of them except Il1β were also significantly increased in mdr2-/- against control livers (Fig. 3B, upper panel). At 6 weeks of age, expression levels of all above named genes were significantly induced in mdr2-/- and car14-/-/mdr2-/- livers, and expression levels of the proinflammatory cytokines Tnfα, Il1β and Mcp1 were significantly higher in car14-/-/mdr2-/- livers compared to mdr2-/- livers (Fig. 3B, middle panels). At 11 weeks of age, expression levels for all above named genes were significantly higher in mdr2-/- livers and car14-/-/mdr2-/- livers, and expression levels for the proinflammatory cytokines Tnfα and Il1β remained significantly higher in car14-/-/ mdr2-/- livers compared to mdr2-/- livers (Fig. 3B, lower panels).
Assessing cytokeratin 19 expression by immunohistochemistry, mRNA and protein expression in the different genotypes at different ages
Immunohistochemistry for cytokeratin 19(Ck19) as a surrogate marker for the ductular reactions was assessed in the liver of 3, 6 and 11 week old mice (Fig. 4A). Occasional larger ducts were stained in 3 weeks old liver, with a prominent ductal wall staining in the in car14-/-/mdr2-/- liver (Fig. 4A, upper panel). More Ck19 positive ducts were detected in the portal triads in car14+/+/mdr2-/- than control liver at 6 weeks of age, and even more in car14-/-/mdr2-/- liver (Fig. 4A, middle panel). At 11 weeks of age, the car14+/+/mdr2-/- liver and in particular the car14-/-/mdr2-/- livers displayed a very striking increase in Ck19 ductal staining in the portal triads (Fig. 4A, lower panels).
Cytokeratin 19 mRNA expression was analyzed in liver lobe 3 of 3, 6 and 11 week old mice (Fig. 4B). The highest expression levels in relation to β-actin was seen in the 3 week old in car14+/+/mdr2+/+, car14-/-/mdr2+/+ and car14+/+/mdr2-/- mice, and at week 6 in car14-/-/mdr2-/- livers, the time point at which it is also significantly higher than in car14+/+/mdr2-/- liver.
Western analysis was performed on the lobe 1 tissue lysate of the mouse liver at week 3, 5 and 11 weeks of age. Vinculin was used as loading control, because it was found to be evenly expressed in different liver segments, and it did not interfere with the size of cytokeratin 19, in contrast to β-actin. Fig. 4C shows representative blots in the left panels, and density ratios of cytokeratin in relation to vinculin in the right panels, in mice aged 3, 6 and 11 weeks. Significant differences in cytokeratin 19 expression between the mdr2+/+ (both car14+/+ and -/-) and the mdr2-/- livers were seen at all-time points, but a significant difference between car14+/+/mdr2-/- and car14-/-/mdr2-/- livers was only seen at week 6 (Fig. 4C, middle panel). At week 6, the car14-/-/mdr2-/- liver displayed also the highest ck19 mRNA as well as Ck19 protein levels in relation to the control gene/loading control at all-time points and in all groups. Please note the different y-axis scaling.
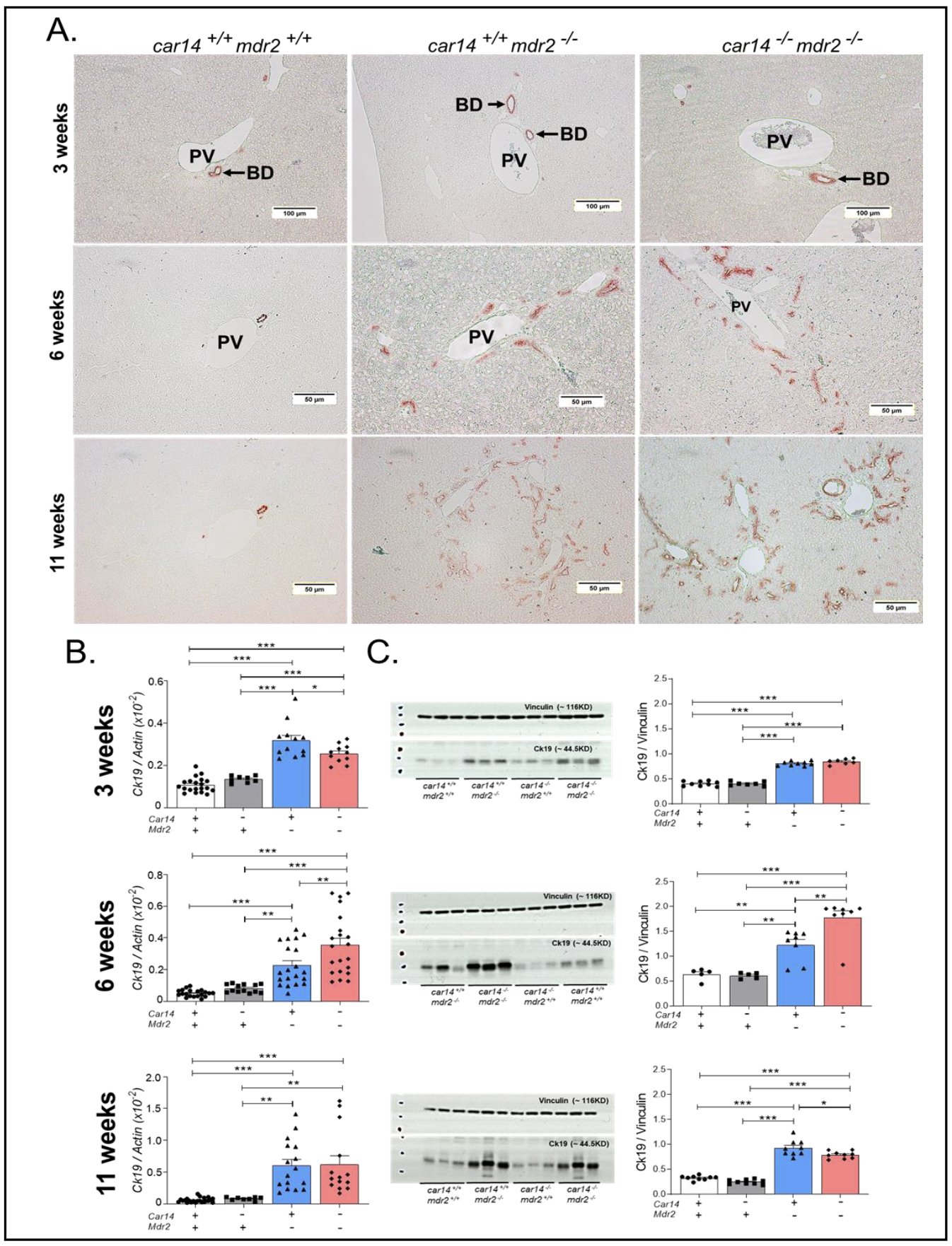
Figure 4. Cytokeratin 19 expression by immunohistochemistry, mRNA and protein expression in the different genotypes at 3,6 and 11 weeks of age. (A) Upper panel: Immunohistochemical staining for cytokeratin 19 in livers from 3 week old mice showed more pronounced ck19 staining (thicker wall) in the car14-/-/mdr2-/- bile ducts, whereas the difference between car14+/+/mdr2-/- and wt littermates was equivocal. Middle panel: Strong increase in the number of ck19-positive bile ducts in the car14+/+/mdr2-/- and more so in car14-/-/mdr2-/- liver at 6 weeks of age. Lower panel: Very strongly increased number of ck19 positive bile ducts in in the car14+/+/mdr2-/- and more so in car14-/-/mdr2-/- liver at 11 weeks of age. (B) CK19 mRNA and (C) protein expression was increased in car14+/+/mdr2-/- and car14-/-/mdr2-/- livers, and the increase was significantly higher in car14-/-/mdr2-/- than the car14+/+/mdr2-/- livers at 6 weeks of age, but not at 3 and 11 weeks of age. Each dot represents a liver sample from one mouse.
Loss of Car14 accelerates the development of fibrosis in mdr2-/- liversNo fibrotic changes were observed in 3 week old mice by Sirius Red staining (Fig. 5A, upper panel), but the Tgfβ2 mRNA expression was already slightly but significantly elevated in the liver of car14+/+/mdr2-/- and car14-/-/mdr2-/- mice (Fig. 5B, right bar graph). At 6 weeks of age, the fibrotic changes were visible around the bile ducts in the car14+/+/mdr2-/- livers, and the car14-/-/mdr2-/- livers also displayed fibrosis between the hepatic lobules (Fig. 5A, middle panels). Collagen formation had become prominent in 11 week old livers, with bridging fibrosis between the portal triads now also in the car14+/+/mdr2-/- livers, and again with more severe pathologic alterations in car14-/-/mdr2-/- than in car14+/+/mdr2-/- livers (Fig. 5A, lower panels). Tgfβ2 mRNA expression had significantly increased car14+/+/mdr2-/- livers, and significantly more so in car14-/-/mdr2-/- livers at 6 weeks of age (Fig. 5B, middle bar graph). At 11 weeks of age, a further increase in the Tgfβ2 mRNA expression had occurred in car14+/+/mdr2-/- livers, now reaching the level of the car14-/-/mdr2-/- livers at week 6, while no change was seen in the control and the car14-/-/mdr2-/- livers.
Overall, the histological and immunohistochemical investigations provide an explanation for the observed decrease in bile flow in car14+/+/mdr2-/- livers between week 6 and week 11, and in car14-/-/mdr2-/- livers at week 6 and week 11, which is likely secondary to the progressive bile duct narrowing and fibrosis observed in these livers during this time span. No pathologic morphological alterations were observed in car14-/-/mdr2+/+ livers at week 3, 6 and 11 (images not different from car14+/+/mdr2+/+ livers, data not shown), although the TUDCA induced HCO3- output was significantly decreased compared to that in car14+/+/mdr2+/+ mice. In contrast, the most severe inflammatory, bile duct proliferative and fibrotic changes in car14-/-/mdr2-/- livers in both 6 week old and 11 week old mice correlated with the lowest bile flow and HCO3- output rates in the four genotypes at each time point. The data demonstrate that deletion of Car14 not only functionally interferes with biliary alkalinity, but also aggravates inflammatory and fibrotic changes in mdr2-/- livers. The inflammatory and cholangiocyte proliferative process appears to reach a plateau, which is reached earlier in the car14-/-/mdr2-/- than in the car14+/+/mdr2-/-livers. We have not investigated the late stages of liver disease with tumor development in these mouse genotypes, however.
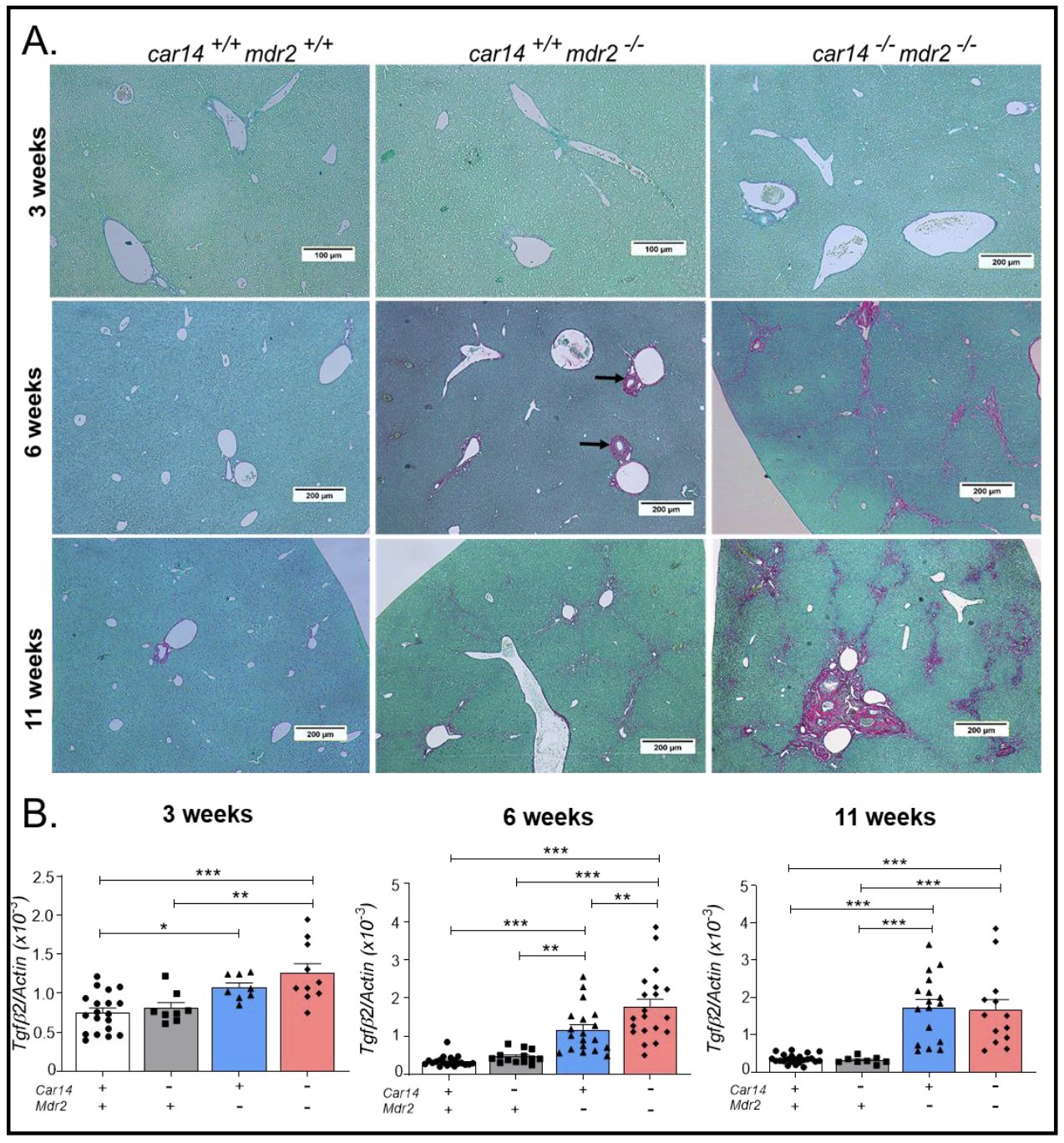
Figure 5. Collagen deposition by Sirius Red staining and expression of profibrotic Tgfβ2 in the liver of the different genotypes at 3,6 and 11 weeks of age. (A) Upper panel: No evidence of fibrotic changes in the three genotypes, Middle panel: Increased collagen staining in the periductal area in the in the car14+/+/mdr2-/- livers, and also “bridging” fibrosis between the portal triads in the car14-/-/mdr2-/- livers. Lower panel: The car14+/+/mdr2-/- livers also show “bridging” fibrosis between the portal triads. Severe periportal fibrotic changes in the portal triad in the car14-/-/mdr2-/- liver. (B) Right bargraph: Significantly higher mRNA expression levels the profibrotic Tgfβ2 in both car14+/+/mdr2-/- and car14-/-/mdr2-/- compared to car14+/+/mdr2+/+ and car14-/-/mdr2+/+ liver tissue in 3 week old mice. Middle bargraph: At 6 weeks of age, the Tgfβ2 mRNA expression had increased strongly in the car14+/+/mdr2-/- and in the car14+/+/mdr2-/- and significantly more so in car14-/-/mdr2-/- liver, while the Tgfβ mRNA expression did not increase relative to β-actin. Lower panel: Tgfβ2 mRNA expression at 11 weeks of age in the four genotypes. Each dot represents a liver sample from one mouse.
Expression of hepatocyte and/or cholangiocyte acid/base transporters, carbonic anhydrases, bile salt and cholesterol transporters, in hepatic tissue from car14+/+/mdr2+/+, car14-/-/mdr2+/+ and car14+/+/mdr2-/- mice.A potential confounding factor for the interpretation of the planned experiments could be a major adaptive change in the expression of the major hepatocyte acid/base and the bile salt transporter in the car14-/- liver and/or the mdr2-/- liver. No change in the hepatic expression of most tested genes was observed between 6 week old car14-/- and wt mice (Fig. 6), except for the following interesting change: while the expression of the predominant intracellular carbonic anhydrase in the liver, namely Car3 (Fig. 6E), was not different between car14-/- and wt liver, the expression level of Car15 (Fig. 6F), another membrane resident carbonic anhydrase with a much lower hepatic expression level than Car14, was significantly upregulated in 6 week old car14-/- liver [40]. These results correspond to the lack of histological alterations and bile flow rates in young and adult car14-/- mice described previously [24].
In contrast, Cftr, Ae2 (Slc4a2) and Asbt (Slc10a2) mRNA expression was significantly upregulated in the mdr2-/- liver at 6 weeks of age (Fig. 6A, B and H). The upregulation of Ae2 may be causative for the increased TUDCA-stimulated HCO3- output at 6 weeks of age, but is an association not a proof. The upregulation of Cftr and Asbt expression is likely a consequence of bile duct hyper-proliferation, because these transporters are expressed in cholangiocytes. At 11 weeks of age, the mdr2-/- mice displayed a downregulation of multiple hepatocyte-expressed proteins, indicating significant damage beyond the biliary tree (Supplementary Fig. 1).
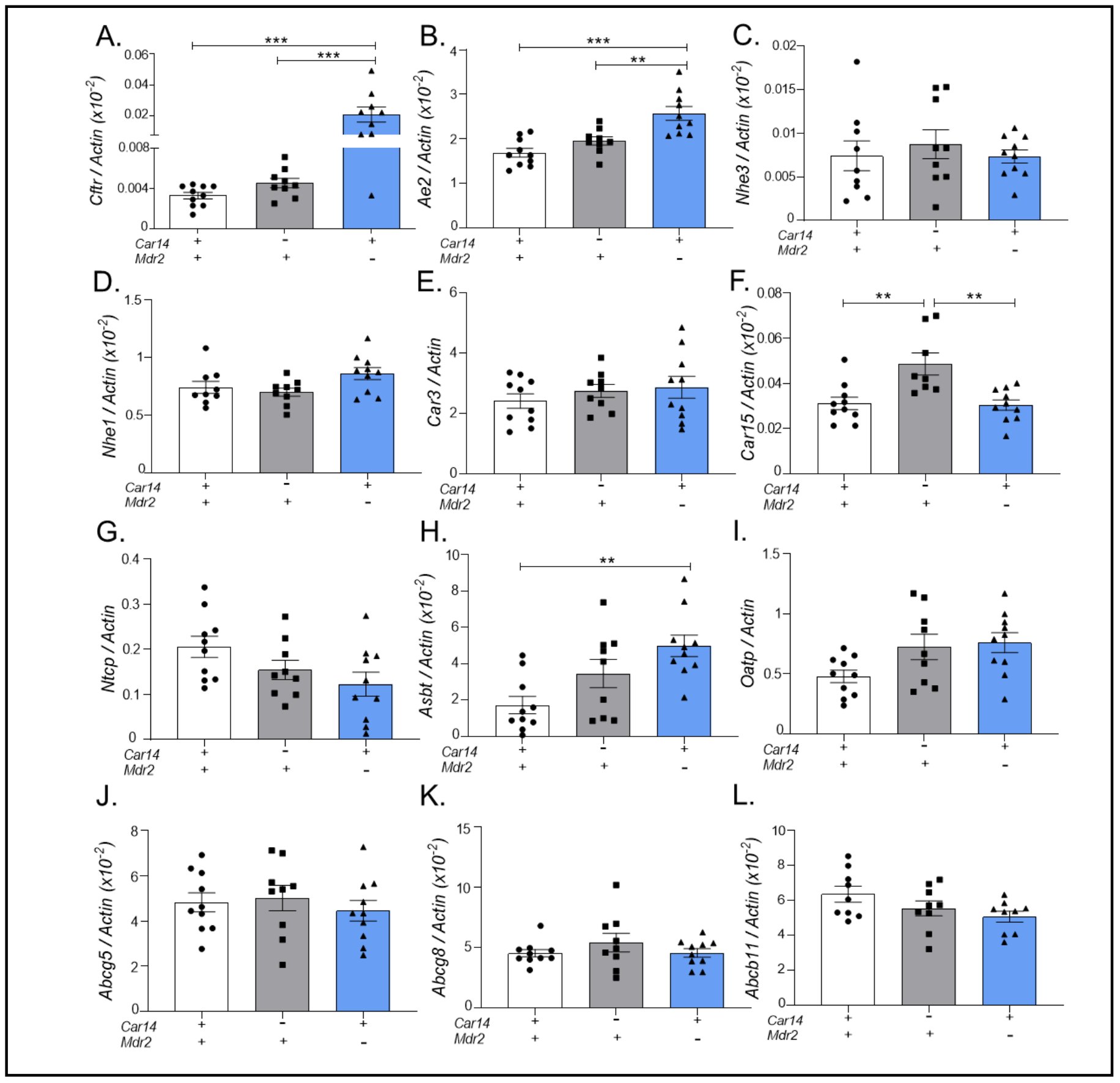
Figure 6. Expression of hepatocyte and/or cholangiocyte acid/base transporters, carbonic anhydrases, and bile salt and cholesterol transporters, in hepatic tissue from car14+/+/mdr2+/+, car14-/-/mdr2+/+ and car14+/+/mdr2-/- mice. The expression of the following genes were assessed by qPCR with β-actin as a control gene: (A) Cftr (Abcc7), (B) Ae2 (Slc4a2), (C) Nhe3 (Slc9a3), (D) Nhe1 (Slc9a1), (E) Car3, (F) Car15, (G) Ntcp (Slc10a1), (H) Asbt (Slc10a2), (I) Oatp (Slco1a1), (J) Abcg5 (K) Abcg8 (Sterolin), and (L) Bsep (Abcb11) in car14+/+/mdr2+/+, car14-/-/mdr2+/+ and car14+/+/mdr2-/- in hepatic tissue of 6 week old mice, to assess adaptive changes in gene expression. In the absence of Car14, the only significant alteration was an upregulation of Car15. In mdr2-/- liver, a significant increase in cholangiocyte-expressed Cftr and Asbt mRNA expression was observed, as well as a mild but significant increase in Ae2 expression. Each dot represents the liver tissue of one mouse. P<0.05 was considered significant.
No difference in the severity of sclerosing cholangiopathy induced by DDC feeding in car14-/- and wt littermatesDDC feeding is an established method for the chemical induction of sclerosing cholangiopathy [33]. In this model, the hepatocellular damage results from protoporphyrin accumulation and the ductal damage from precipitation of the poorly soluble protoprophyrin in the ducts, followed by intraductal stasis, ruptural and periductal inflammation. Three week feeding of 0.1% DDC supplemented diet resulted in progressive and similar weight loss in the third week in both car14-/- and wt mice by 11 0.4% of the initial weight (n=7). Mice that had lost <30% of their initial body weight were excluded from the analysis, because the feeding regimen had been stopped and the mice were sacrificed as per approved animal experimentation protocol. Bile flow and biliary HCO3- secretion was assessed and is shown in Fig. 7A, B. A significant reduction in basal and TUDCA stimulated bile flow was observed in DDC fed, car14-/- and wt mice, compared to wtcontrols (Fig. 7A). Likewise, a severe reduction in biliary HCO3- output was observed in DDC fed mice in the basal state and after TUDCA stimulation, which was even lower in DDC fed car14-/- than wt mice (Fig. 7B). The liver had obtained a dark colour and a stiff texture (Fig. 7C), and a severe sclerosing cholangiopathy was observed in both DDC fed car14-/- and wt mice (Fig. 7D). No obvious difference was seen in the severity of damage between DDC fed car14-/- and wt mice. Cytokeratin19 expression was strongly increased in liver tissue of DDC treated mice, but there was no difference between car14-/- and wt livers (Fig. 7E).
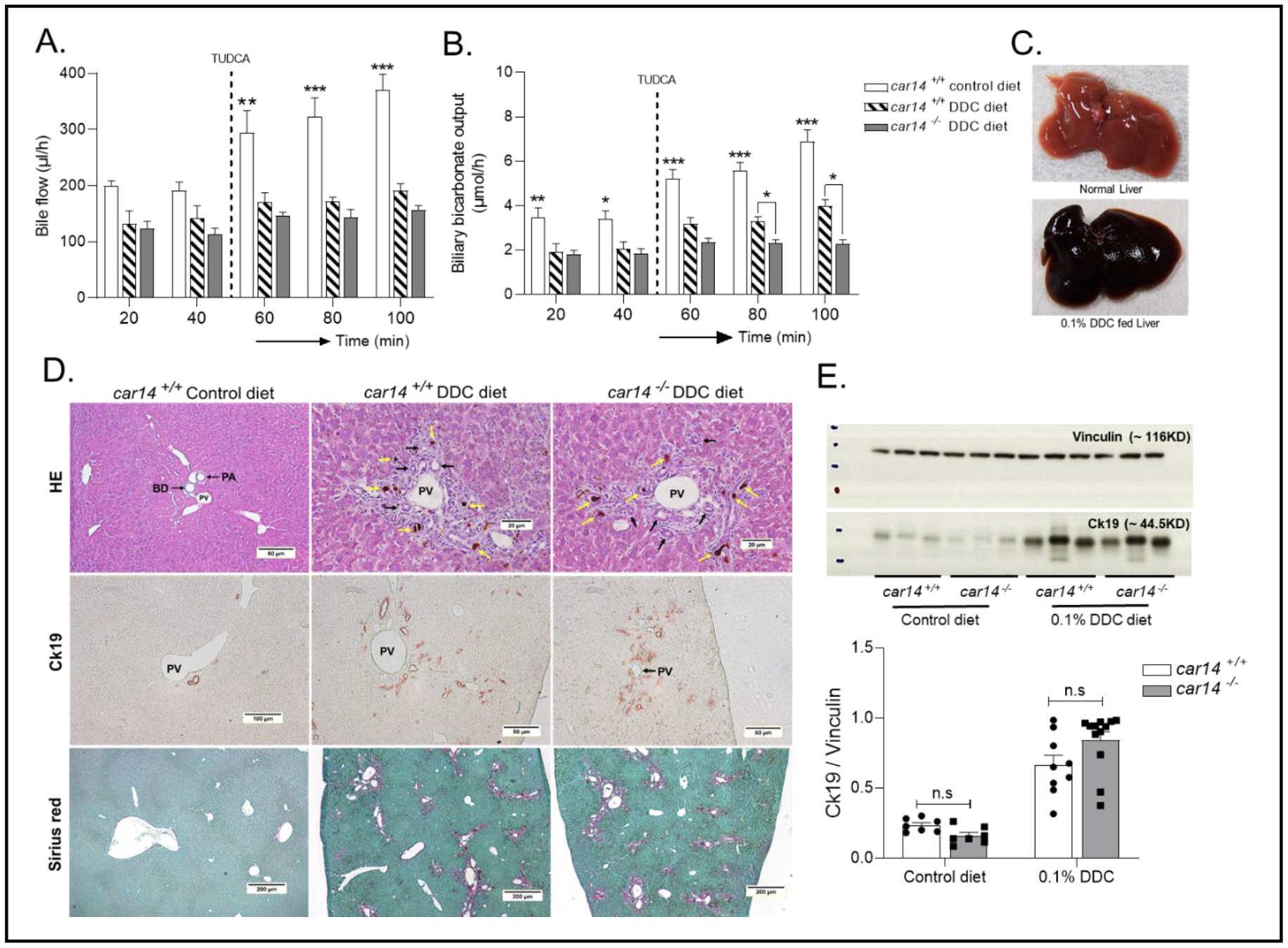
Figure 7. Functional, histopathologic and cholangiocyte proliferative alterations in the car14-/- and wt mice after DDC feeding. (A) Bile flow rates in control wt mice, DDC fed wt mice and DDC fed car14-/- mice. After 3 weeks of DDC (0.1% wt/wt in the food), bile flow rates were significantly reduced compared to chow-fed littermates in both car14-/- and wt littermates. (B) Hepatobiliary HCO3- output rates in control wt mice, DDC fed wt mice and DDC-fed car14-/- mice. DDC feeding resulted in significantly reduced hepatobiliary HCO3- output in both wt and car14-/- mice, with significantly lower HCO3- output rates in the latter. (C) Dark colour and stiff texture of the liver after 0.1% DDC supplemented diet for a period of 3 weeks. (D) Histopathological appearance of the DDC-fed liver: H&E stain demonstrating the thickening and immune cell infiltration of the large bile ducts (upper panels), Cytokeratin 19 (ck19) immunostaining demonstrated the cholangiocyte proliferation, and Sirius red staining uncovers the development of liver fibrosis. No differences were seen between DDC fed wt and car14-/- mice. (E) Western analysis revealed strong increase in ck19 expression in DDC fed wt and car14-/- mice. n=7-8 for control diet-fed wt and car14-/- mice, 9-11 for DDC fed wt and car14-/- mice.
Carbonic anhydrase XIV (CAXIV, Car14) is the most recently identified CA with predominant expression in brain, kidney, liver, skeletal muscle, heart, and lung [41]. Car14 is highly expressed in hepatocytes with predominance in the canalicular membrane, supposedly with its active site in the extracellular (luminal) milieu [42]. Membrane bound carbonic anhydrases have been shown to augment the dissipation of pH-gradients and thus increase the speed of acid or base transport via the membrane [24, 26, 27]. In a previous report, we had measured basal and TUDCA stimulated hepatic bile flow and HCO3- secretory rates in car14-/- mice and wt mice whose common bile duct was cannulated [24]. Indeed, a significantly decreased alkaline output into bile was found, but no difference in basal or TUDCA stimulated bile flow, no histological alterations and no increase in hepatic proinflammatory, bile duct proliferative and profibrotic gene expression at young and middle age. In this study, we complemented the data by assessing the expression of a panel of hepatic acid/base, bile salt, cholesterol transporters, and liver expressed carbonic anhydrases (Fig. 6). The only significant difference was an approx. twofold upregulation of Car15, the only other membrane bound hepatic anhydrase. However, given its low expression level compared to Car14, this is unlikely to compensate for much of the lost Car14 function.
Therefore, a mouse model was selected which displays a more hydrophobic, toxic bile acid pool, and generate a mouse with a more toxic bile acid pool and a selective reduction of biliary HCO3- output in conjunction with an unaltered bile flow rate. One such model might have been the Cyp2c70 knockout mouse, which lacks the enzyme that converts chenodeoxycholic acid (CDCA) to hydrophilic muricholic acids (MCAs), and develops hepatic inflammation as a consequence [43]. This model was not available to us. Another model is the mdr2-/-mouse model of sclerosing cholangitis. As mentioned in the introduction, the absence of the canalicular phospholipid transporter ABCB4 (MDR3, mouse homologue Mdr2) results in a sclerosing cholangiopathy in the mdr2-/- mouse. The lack of phospholipids in the bile impedes the formation of “mixed micelles” and “liquid crystals”, in which the water-insoluble cholesterol is kept in solution by bile acids and phospholipids, mostly lecithin, for transport into the intestine [44-47]. In the absence of ABCB4 (Mdr2), an excess of mostly hydrophobic bile acids is present in the hepatic bile and causes bile duct injury, leakage, and inflammation, ultimately resulting in a sclerosing cholangiopathy, followed by biliary cirrhosis and the development of inflammation-induced hepatocellular carcinoma [1, 48]. Mutations of ABCB4 in humans likewise cause a variety of cholestatic liver diseases, including sclerosing cholangiopathy [49, 50]. The feeding of hydrophilic bile acids to mdr2-/- mice ameliorates the hepatobililary damage [16, 51].
Deletion of Car14 in mdr2-/- mice appears to be therefore a good strategy to study the protective effect of biliary HCO3- output against bile acid toxicity in an animal model. We examined Car14 expression in mdr2-/- and wt liver at 6 and 11 weeks, and observed a robust Car14 expression in mice with cholestatic liver injury. By breeding car14-/-/mdr2-/- double deficient mice, the protective effect of Car14 expression was evaluated by comparison with the mdr2-/- liver phenotype in respect to biliary HCO3- output, bile flow, liver histopathology and the expression of inflammatory, cholangiocyte proliferative and profibrotic markers. Although we noticed that the histological alterations were stronger in the liver periphery, similar to what has been reported in the original description of the phenotype of the mdr2-/-mouse model [52], we used whole liver lobes for mRNA and protein expression, although this may underestimate the pathologic changes. The mice were studied until 11 weeks of age, which was the time span during which no weight loss, no change in feeding pattern or signs of well-being and no hepatic malignancies develop, which are all hallmarks of this mouse model at later stages [52]. The protective effect of Car14, assessed by the significant difference in inflammatory, cholangiocyte proliferation, and profibrotic markers between car14+/+/mdr2-/- and car14-/-/mdr2-/- liver, was most consistently seen at 6 weeks of age. At that time point, the difference in biliary HCO3- output was very high between car14+/+/mdr2-/- and car14-/-/mdr2-/- mice. In addition, a significantly higher HCO3- output was observed in the car14+/+/mdr2-/- compared to the car14+/+/mdr2+/+ (wt) mice (Fig. 2), possibly (but not proven) related to the upregulation of Ae2 (Fig. 6B) and Car14 at 6 weeks of age (Fig. 1), but not at 11 weeks of age, possibly due to progressive hepatocyte damage (Supplementary Fig. 1).
The results support the hypothesis generated in a previous publication in which the dual farnesoid (FXR/TGR5) receptor agonist INT-767 (which is a hydrophilic bile acid analogue) was applied to mdr2-/- mice [53]. The drug caused a strong decrease in bile acid synthesis, an increase of HCO3- rich choleresis, with presumed hepatocyte origin, and an upregulation of Car14 expression [53]. A hypothetical model was created that explains both the increased bile flow and the biliary HCO3- output by a transport metabolome of apical AE2 and CAR14 in the hepatocyte canalicular membrane. The speculated importance of Car14 for the generation of HCO3- rich bile fluid is confirmed by our study, but in contrast to the above study, our investigations were able to separate the (well-known) protective effect of an increased bile flow from that of the (hypothetical) protective effect of biliary HCO3- output.
The relatively small (and not always significant for each studied parameter in each age group) difference between the car14+/+/mdr2-/-and the car14-/-/mdr2-/- livers in this study is in stark contrast with the dramatic differences in post bile duct ligation damage between the livers from Car14 knockout mice, bought from Cyagen where they were created by Crispr/Cas genome editing, and wt livers from mice raised in the animal facility of Zheijang University [54]. The most conspicuous difference after 7 day bile duct ligation was the appearance of massive necrotic areas in the liver of the Car14 knockout mice, which were not visible in the wt livers. Parenchymal necrosis is a feature of the bile duct ligation model, and is due to acute and massive bile salt retention in the hepatocyte [55]. Such a dramatic difference in degree of necrosis in the presence or absence of a membrane bound carbonic anhydrase would be surprising, and may point to an increased vulnerability of the Car14 knockout livers due to other reasons than the absence of Car14. Since the Car14 knockout and the wt mice were from different pedigrees, off target effects of the Crispr/Cas technology, which are a well-known problem [56, 57], genetic drifts during inbreeding with homozygotes, and a different gut microbiome due to the different origin of the Car14 and the wt mice [58], may have contributed to both the extreme vulnerability of the Car14 knockout, as well as the robustness of the wt mice to this very severe, multi-organ damage model [59, 60].
To control for a potential influence of Car14 on protective functions unrelated to the biliary HCO3- output, we tested the effect of Car14 deficiency in another mouse model of sclerosing cholangiopathy. The drug DDC inhibits the mitochondrial enzyme ferrochelatase, leading eventually to a progressive accumulation of protoporphyrin. The hydrophobic protoporphyrin that accumulates in hepatocytes and Kupffer cells, is excreted into bile where it is poorly soluble and causes occlusion of the small bile ducts with subsequent inflammation [29, 30, 61]. For this model, a role of biliary HCO3- is unlikely. However, it cannot be ruled out that Car14 augmented intracellular pHiregulation influences the response to toxic injury unrelated to bile acid injury to the ductal system. DDC feeding resulted in periductular sclerosis and infiltration, porphyrin plugs, and fibrosis, but the absence of Car14 did not cause significant aggravation of CK19 upregulation, or a visual difference in the morphological phenotype, suggesting that the hepatocyte acid/base balance and intraductular HCO3- concentrations does not play a role in DDC induced liver damage.
In summary, the deletion of the carbonic anhydrase 14 in the mdr2-/- mouse model aggravated the cholestatic liver injury that develops in this mouse model due to the presence of an excess of bile acid monomers. The cartoon in Fig. 8 displays key aspects of the molecular pathology important in causing the hepatocholangiocellular damage seen in the car14-/-/mdr2-/- liver. The car14-/-/mdr2-/- mouse model is to our knowledge the first animal model that demonstrates the important role of hepatocyte HCO3- output to protect the biliary system against bile acid damage. These results suggest that systemic acid/base balance may be of critical importance in patients at risk for biliary disease, and warns against the uncritical use of the many drugs with carbonic anhydrase inhibitory potential in the presence of liver disease.
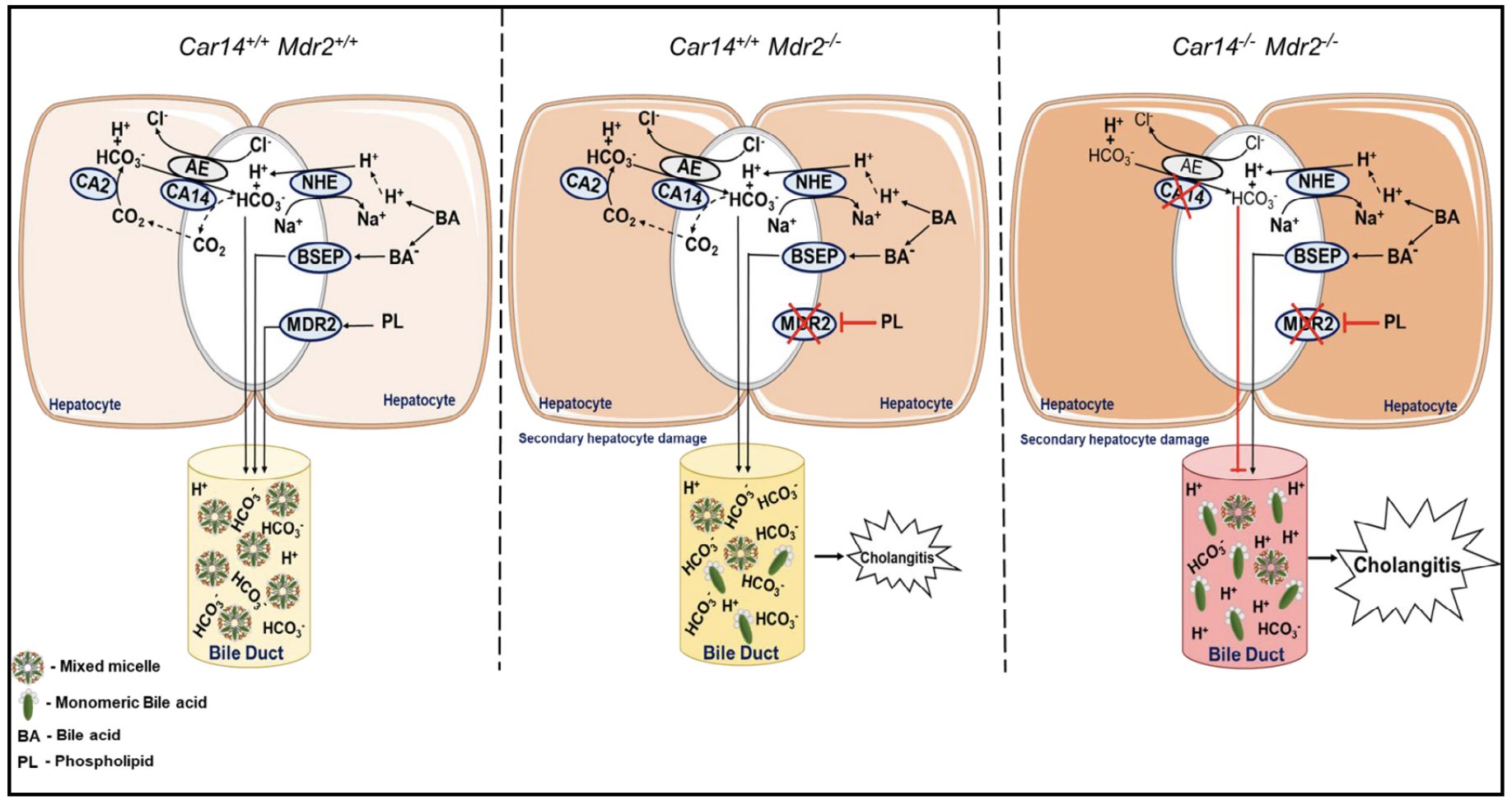
Figure 8. Cartoon depicting the potential role of CAXIV in the protection of the biliary tree against bile acid toxicity. Left cartoon: In health, the MDR2 mediated transport of lecithin and the AE2 mediated, Car14-augmented export of HCO3- into the canaliculi allow for a complete integration of the BSEP excreted bile acid monomers into mixed micelles containing bile acids, lecithin and cholesterol. Middle panel: In the absence of MDR2, phospholipid transport into bile is reduced, the formation of mixed micelles is impaired, resulting in an increase of monomeric bile acids, which are damaging to the biliary epithelium. Possibly as a compensatory mechanism, the biliary HCO3- concentration is increased in young mdr2-/- mice, Ae2 and Car14 mRNA are upregulated, and bile flow is maintained despite initiation of periductal inflammation. In adulthood, bile flow as well as HCO3- output are reduced, inflammation, cholangiocyte proliferation and fibrosis severe, and hepatocyte transporter gene expression reduced, indicative of both bile ductal and parenchymal damage. Right panel: In the absence of CAR14 and MDR2, the HCO3- output is reduced even at young age. The inflammatory, cholangiocyte proliferative and fibrosing changes in the liver of car14-/-/mdr2-/- mice are accelerated.
We sincerely thank all members of the Institute of Animal Research for helping with the complicated breeding strategy. We further sincerely thank Dr. Taolang Li and Dr. Jiajie Qian for establishing the bile sampling technique and contributing data to the manuscript during their thesis work in our group.
Author contributions
A. K., Z.Z., B.R. and U.S. designed the experiments, Z.Z, A.K., A.S., B.R, and D.R. performed and analysed experiments, B.R and D.R. planned the breeding, raised and genotyped the mice, US wrote the manuscript, A.K., U.S. and A.V. revised the manuscript, A.V. provided the mdr2-/- mouse strain.
Financial supportThe work was funded by the DFG grants SE 460/19-1, 21-1 and 22-1 and the Volkswagen Foundation grant Z1953
Statement of Ethics
All animal experiments were approved by the Local Institutional Animal Care and Research Advisory Committee at the Hannover Medical School and authorized by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES) (TVA Nr. 33-12-42502-04-15-1847).
The authors have no conflicts of interest to declare.